Analyse de données métagénomiques shotgun
Module 24
Olivier Rué 
MaIAGE
Valentin Loux
MaIAGE
Cédric Midoux
PROSE & MaIAGE
June 5, 2023
Introduction
Practical informations
- 9h00 - 17h00
- 2 breaks morning and afternoon
- Lunch at INRAE restaurant (not mandatory)
- Questions allowed
- Everyone has something to learn from each other
Better knwow you
Who are you?
- Institution / Laboratory / position
What is your scientific topic?
- Studied ecosystem
- Scientific question
- Experimental design
What is your background?
- Already treated shotgun data?
- Background in bioinformatics?
Better know us

- Open infrastructure dedicated to life sciences
- Computing resources, tools, databanks…
- Dissemination of expertise in bioinformatics
- Design and development of applications
- Data analysis
Data analysis service
- We are specialized in genomics/metagenomics
- Bioinformaticians and Statisticians
- More than 110 projects since 2016
- 2 types of services
- Collaboration or Accompaniement
Training objectives
- Know the advantages and limits of shotgun metagatenomics data analysis
- Identify tools to analyze your data
- Run tools with migale resources
Program
Day 1
- Introduction
- Reminders
- QC
- Taxonomic classification
Day 2
- Cleaning
- Assembly
- Annotation
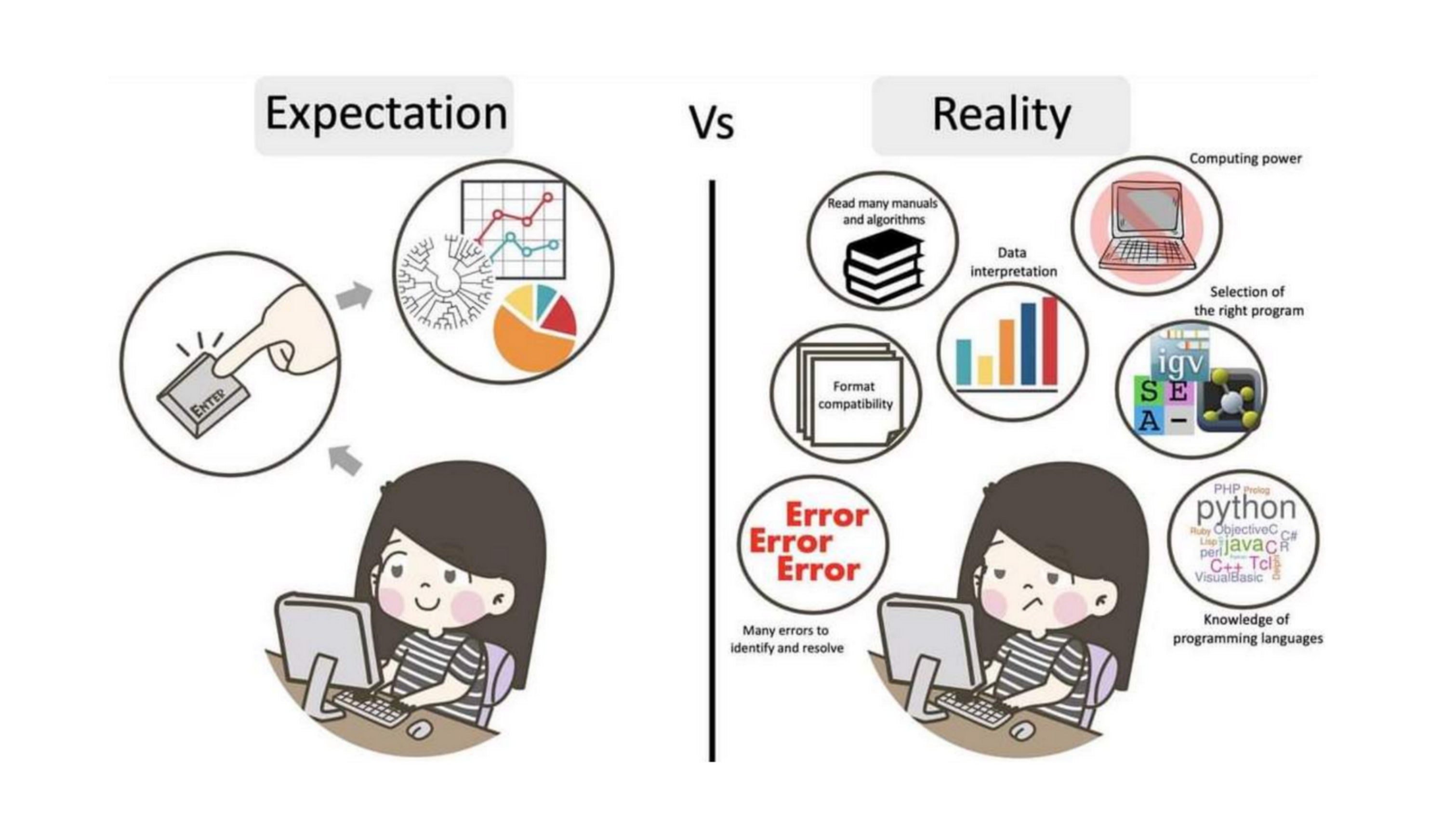
The truth about bioinformatics
Introduction to metagenomics analyses
Introduction
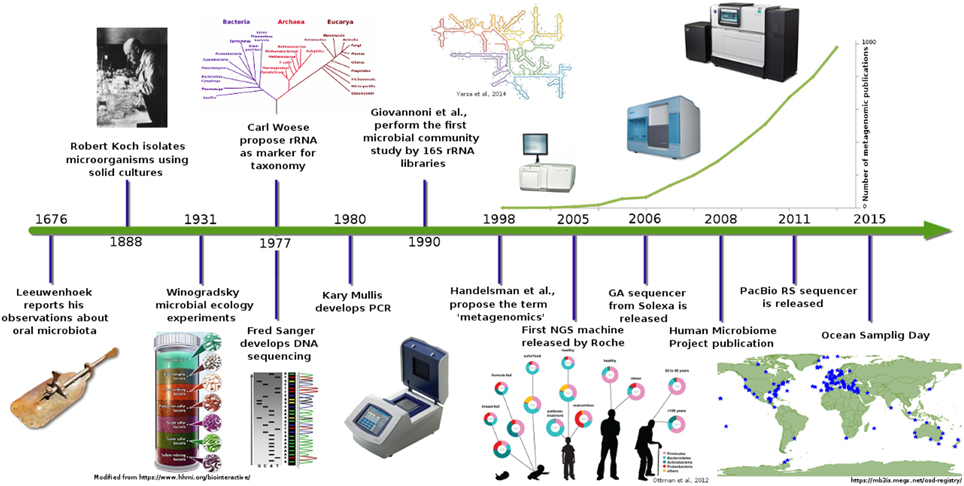
Introduction
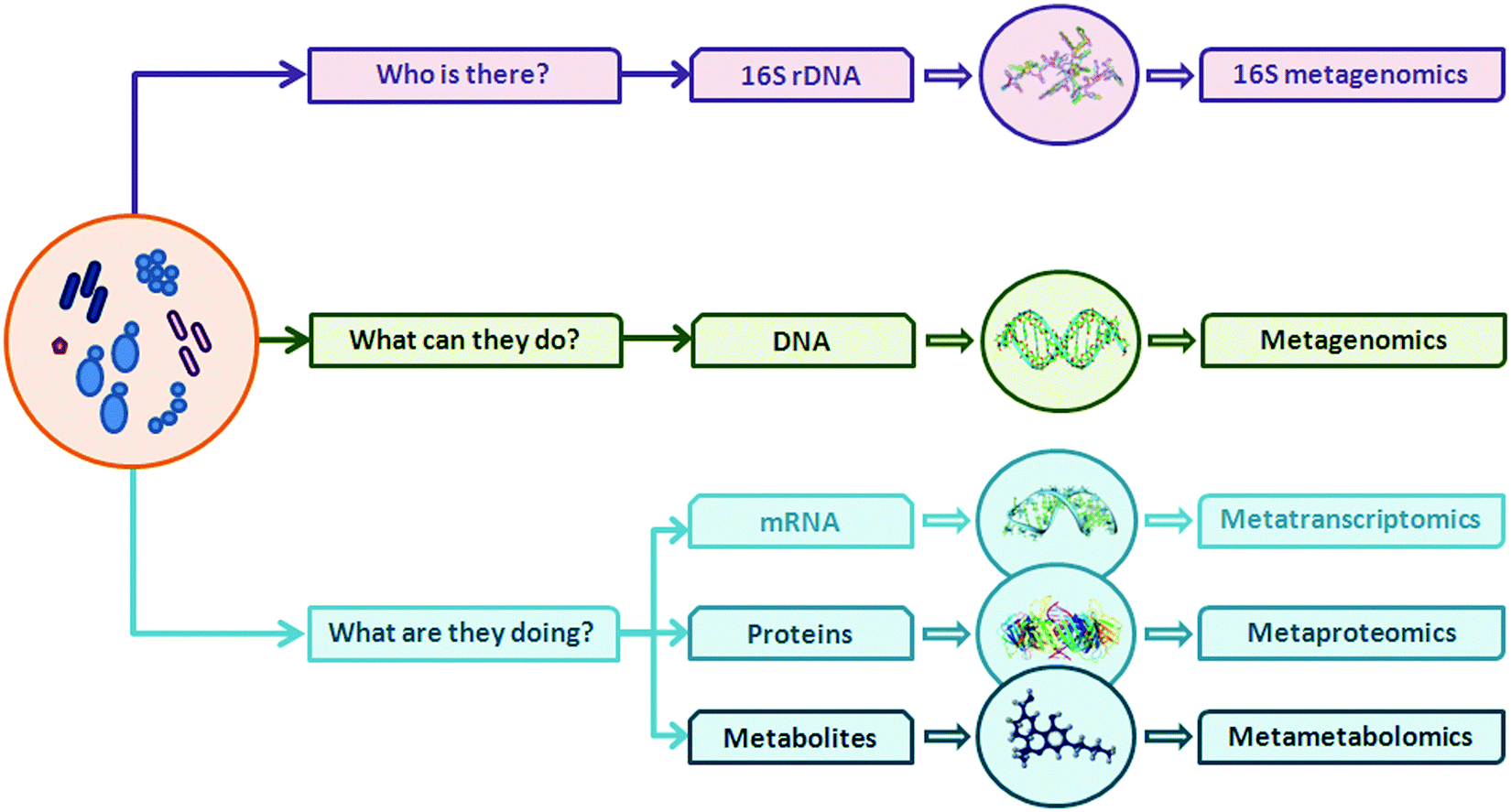
Introduction

Challenges
- Complexity of the ecosystem
- Completeness of databases
- Sequencing depth
- Computational resources required
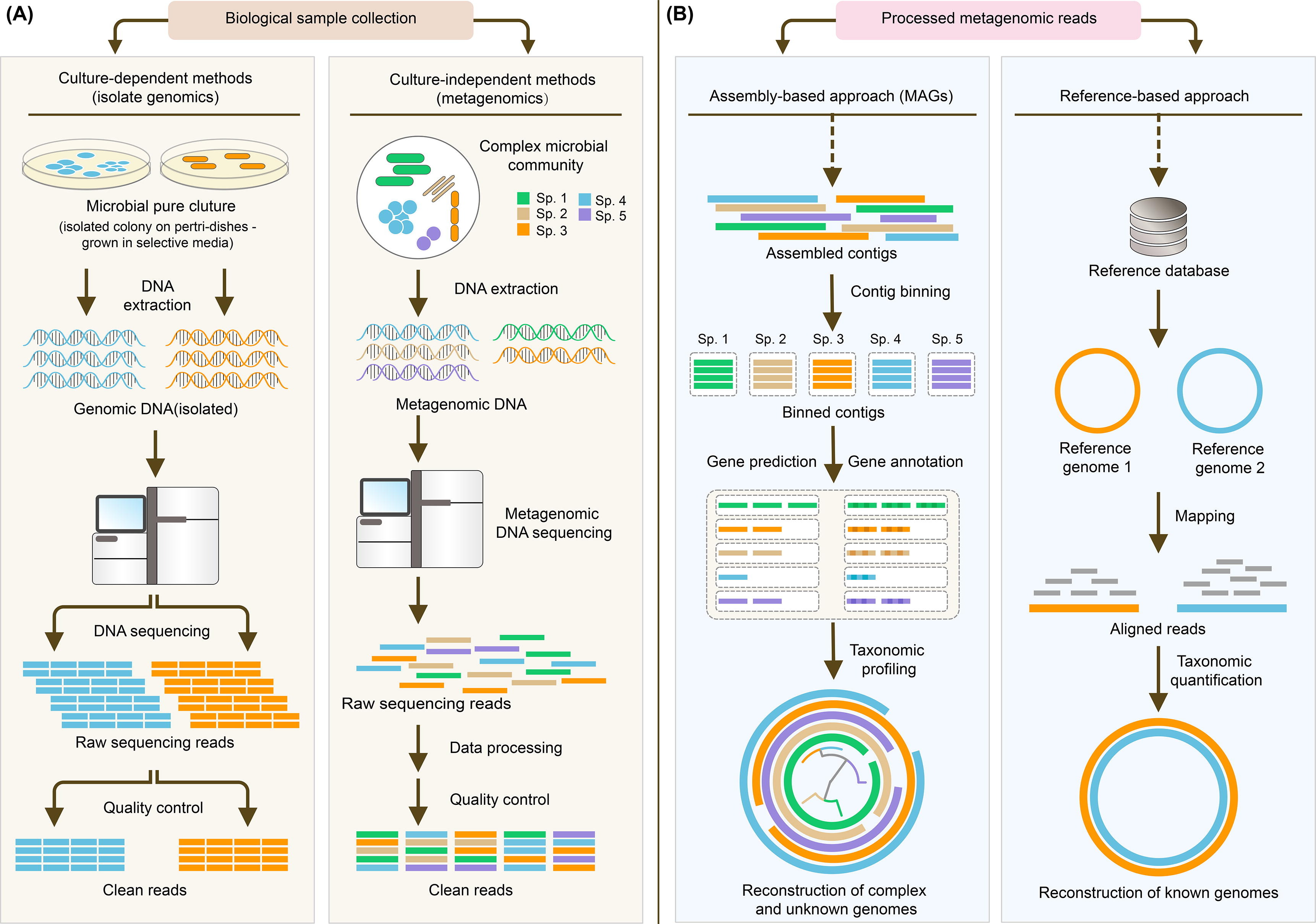
Classical metagenomics approach
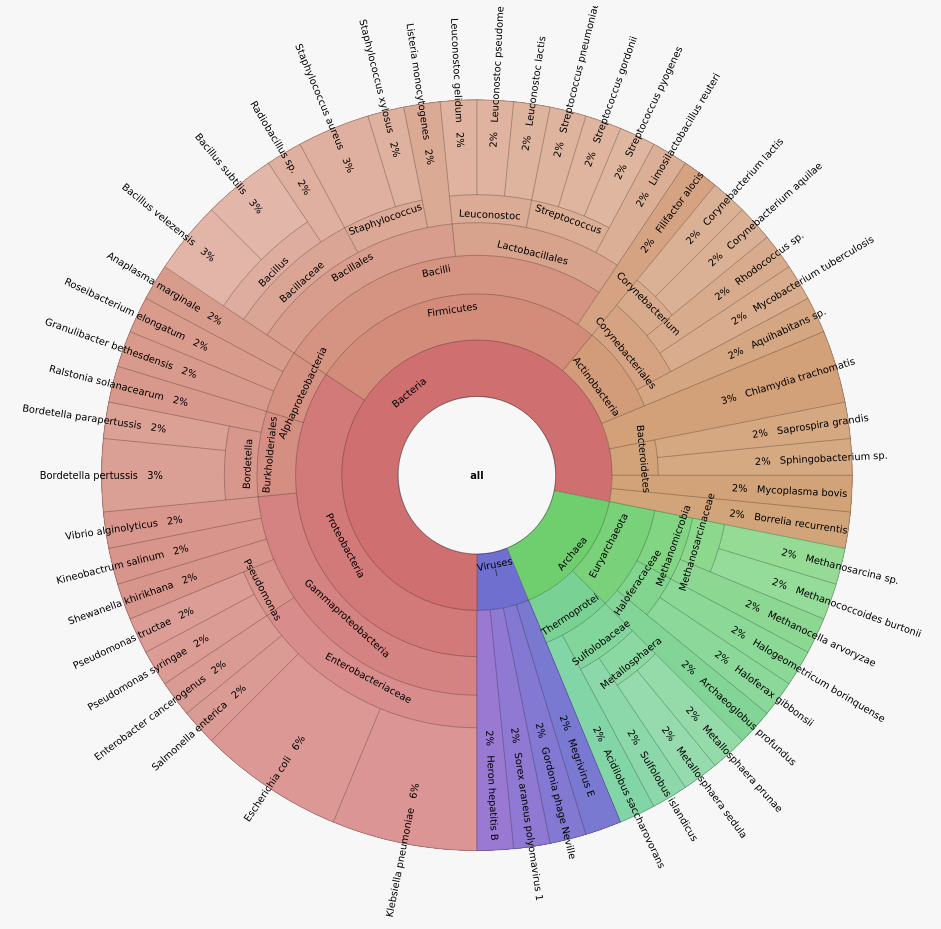
Toy datastet presentation
Home-made dataset
- 5 samples
- 1M or 2M reads per sample
- 50 Bacteria, 10 Archaea, 4 Viruses
- Illumina Hiseq 2x125 bp
- Uniform or log-normal abundance
conda activate insilicoseq-1.5.4
# après un premier tirage aléatoire permettant de récupérer 50 génomes bactériens, 4 génomes viraux et 10 génomes d'archées
iss generate --seed 13062022 -k bacteria viruses archaea -U 50 4 10 --model hiseq --output s1 -n 2000000 --abundance_file hiseq_ncbi_abundance.txt
iss generate --seed 14062022 --genomes s1_genomes.fasta --model hiseq --output s1 -n 2000000 --abundance_file s1_abundance.txt -z
iss generate --seed 14062022 --genomes s2_genomes.fasta --model hiseq --output s2 -n 2000000 --abundance_file s2_abundance.txt -z
iss generate --seed 14062022 --genomes s1_genomes.fasta --model hiseq --abundance uniform --output s3 -n 2000000 -z
iss generate --seed 14062022 --genomes s2_genomes.fasta --model hiseq --abundance uniform --output s4 -n 1000000 -z
iss generate --seed 14062022 --genomes s1_genomes.fasta --model hiseq --output s5 -n 2000000 --abundance_file s1_abundance.txt -zDataset composition
Reminders about command line
The migale facility
- working directories (
save/work/home) - SGE submission system (
qsub/qstat) - conda environments (
conda activate)
All usefull information

Switch to TP 1
Reminders about sequencing
Generations
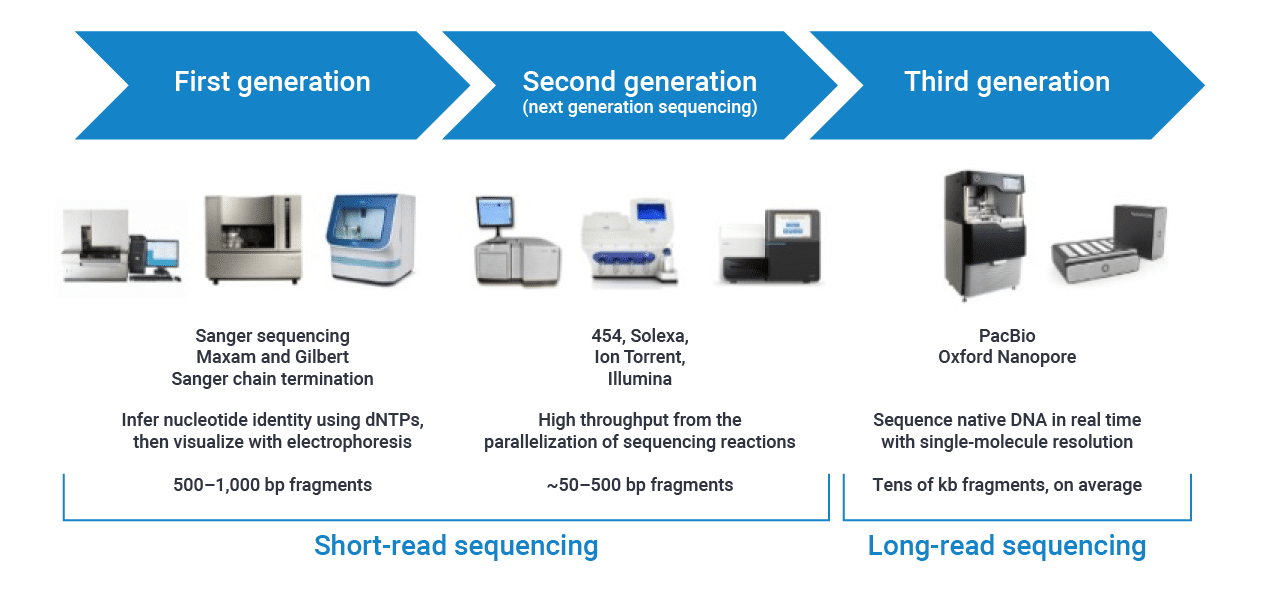
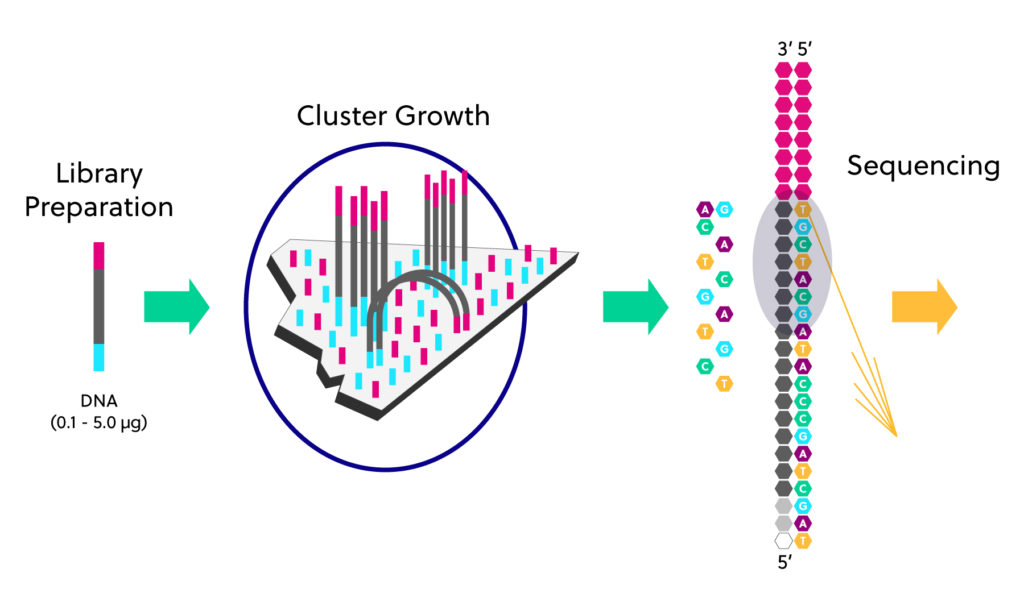
Reminders about Illumina sequencing
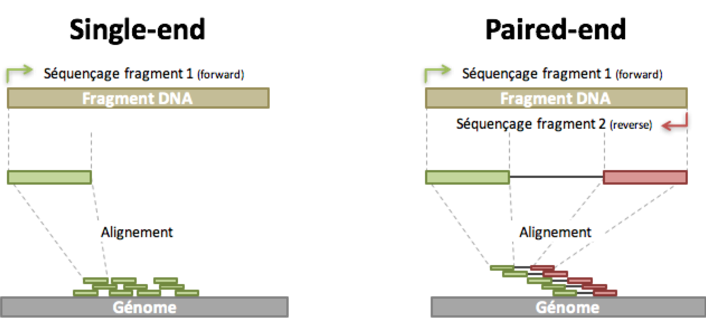
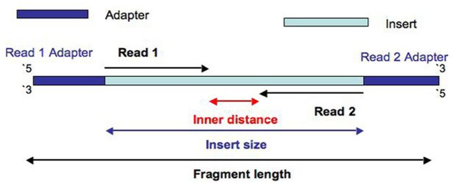
Sequencing - Vocabulary
- Read: piece of sequenced DNA
- DNA fragment: 1 or more reads depending on whether the sequencing is single- or paired-end
- Insert: Fragment size
- Depth: \(\frac{N * L}{G}\)
\(N =\) number of reads
\(L =\) reads size
\(G =\) genome size - Coverage: % of genome covered

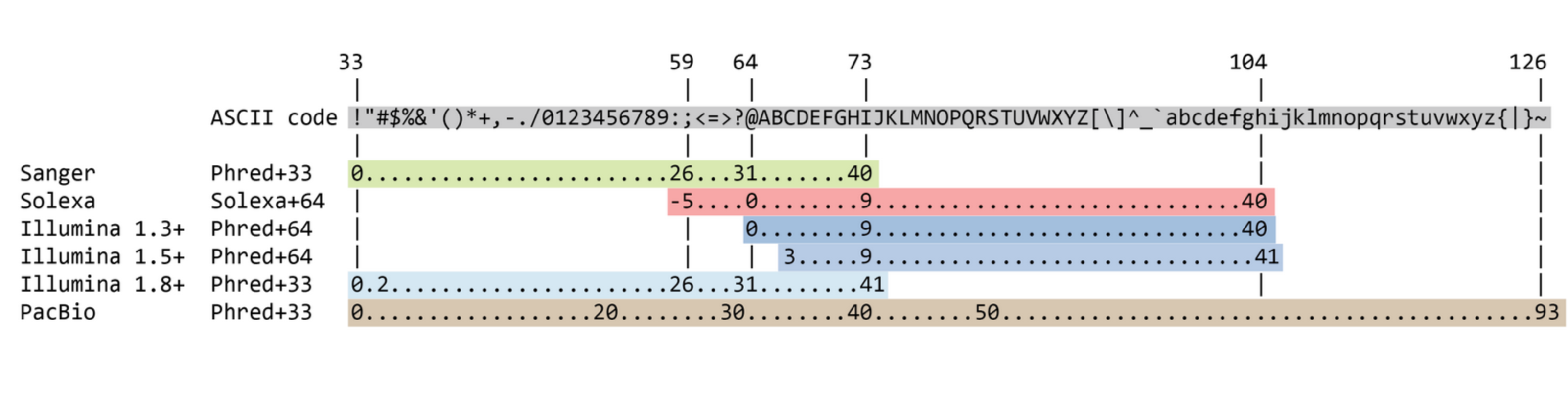
FASTQ format
Meaning
@Identifier1 (comment)
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
+
QQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQ
@Identifier2 (comment)
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
+
QQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQuality score encoding
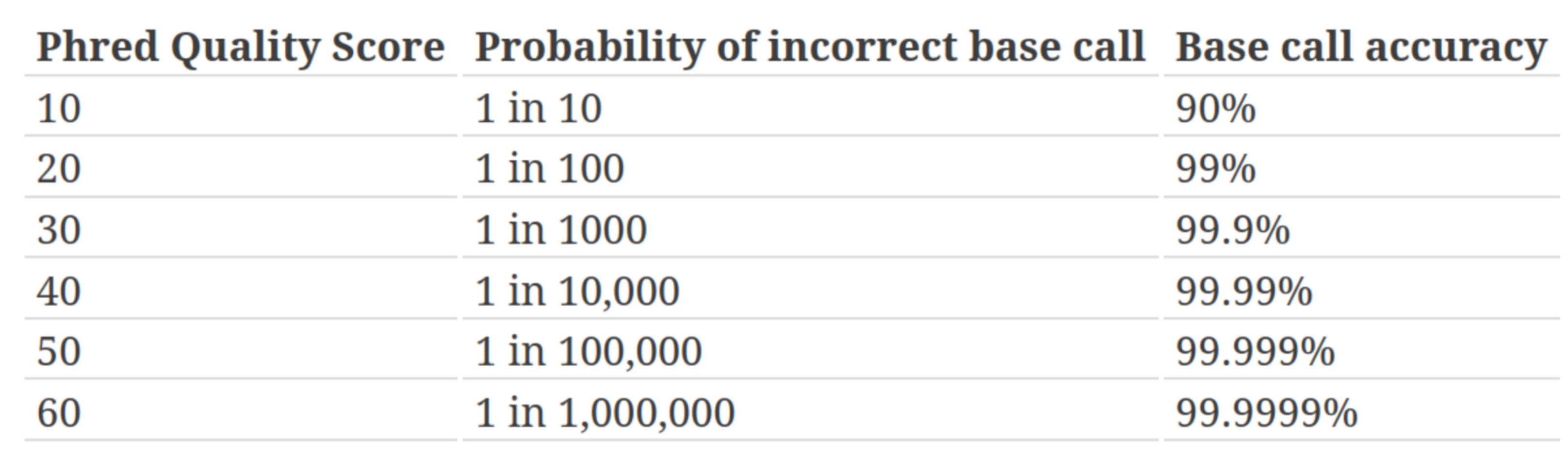
Quality score
Measure of the quality of the identification of the nucleobases generated by automated DNA sequencing
FASTQ compression
- Compression is essential to deal with FASTQ files (reduce disk storage)
- extension:
file.fastq.gz - Tools are (almost all) able to deal with compressed files
Quality control
Quality control
- One of the most easy step in bioinformatics …
- … but one of the most important
- check if everything is ok
- Indicates if/how to clean reads
- Shows possible sequencing problems
- The results must be interpreted in relation to what has been sequenced
Reads are not perfect
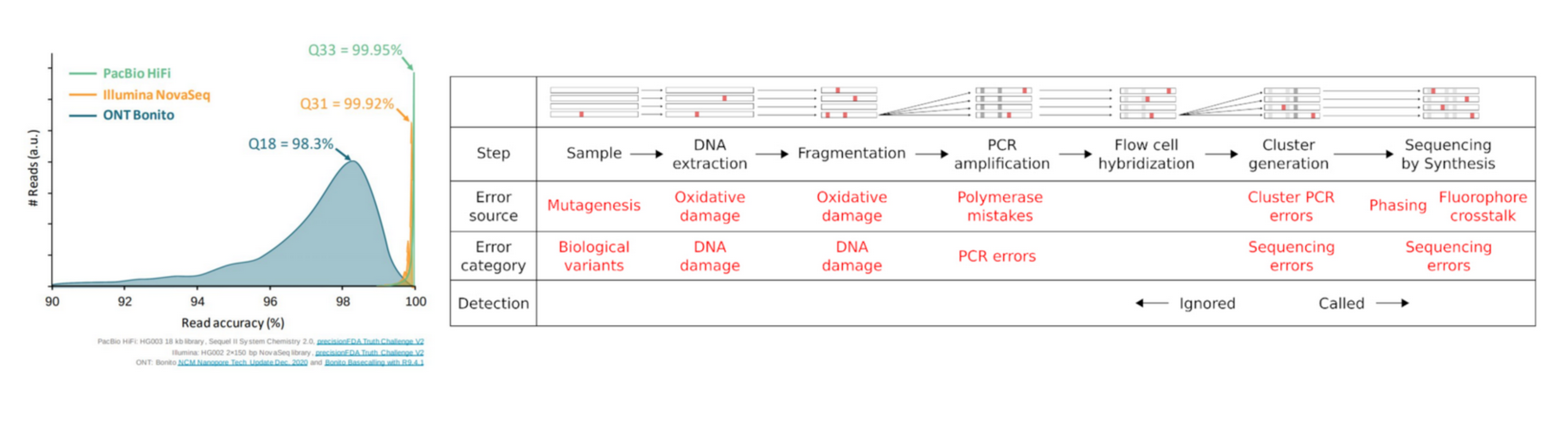
Why QC’ing your reads?
Try to answer to (not always) simple questions:
- Are data conform to the expected level of performance?
- Size / Number of reads / Quality
- Residual presence of adapters or indexes?
- (Un)expected techincal biases?
- (Un)expected biological biases?
Warning
QC without context leads to misinterpretation!
Switch to TP 2
Taxonomic affiliation from raw reads
Kraken
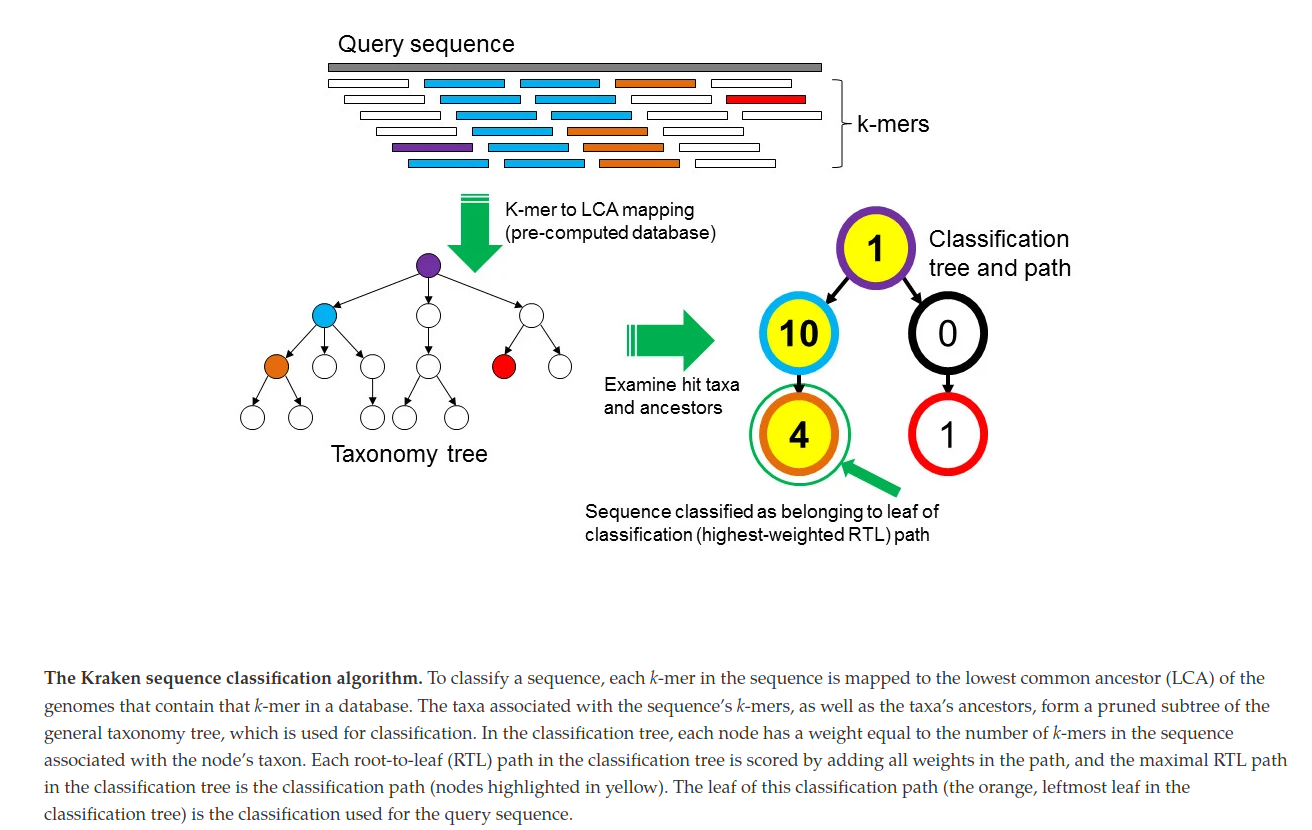
Kraken [5]
- Chop genomes into k-mers and link to a taxonomic id.
- Chop reads into k-mers and search for exact hits in database
- Search for highest-wighted root-to-leaf paths and assign the taxonomic id of the lowest node to read
Kraken
Kaiju
- Database of proteic sequences
- Supposed to be more sensitive
- Translate reads in all six reading frames, split at stop codons
- Use BWT and FM-index table
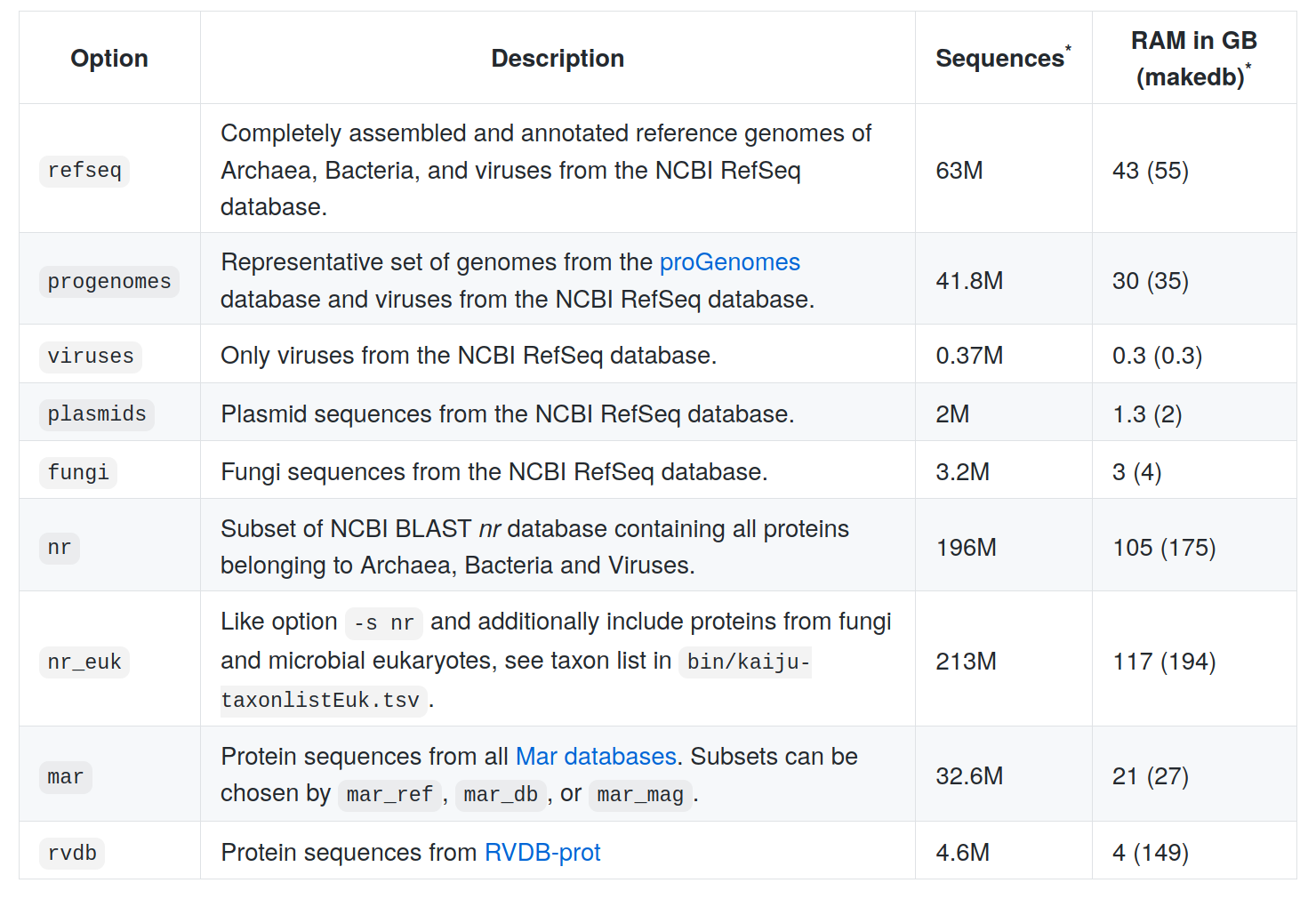
Kaiju databanks
Taxonomic classification caveats
- Databanks
- K-mer choice (sensitivity / specificity)
- Allow a “fast” overview of your data
- Contaminants?
- Host reads?
- Classification rate
Switch to TP 3
Reads cleaning
Quality/Length/Adapters
- Detect and remove sequencing adapters present in the FASTQ files
- Filter / trim reads according to quality (seen with FastQC)
rRNA removal
- Mandatory step if you want to skip rRNA reads
Contamination
- Contamination refers to DNA within a sample that did not originate from that specific biological sample
- Recognizing contamination, followed by appropriate decontamination, should be a critical first step for all microbiome analyses.
- Skipping this step can easily result in confounded results and false conclusions.
Contamination origins
- External contaminants
- Microbial DNA from the surrounding environment
- native microbiomes of researchers
- microbial DNA present in DNA extraction and library preparation kits
Note
Negative controls enable to detect these contaminants
- Cross-sample contaminants
Note
More difficult to assess
Switch to TP 4
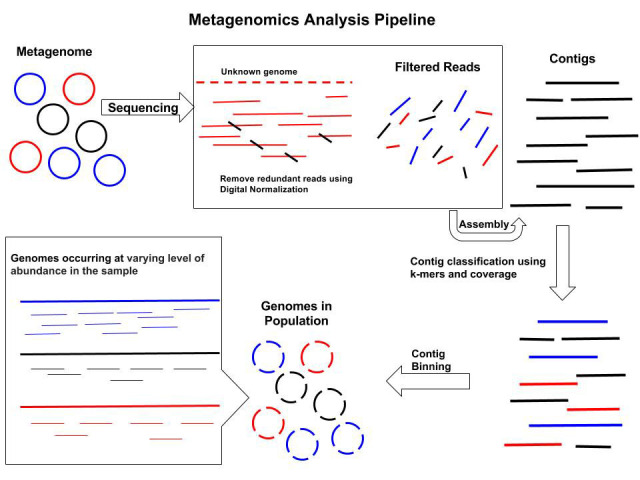
Metagenomics assembly
Objectives
- Reconstruct genes and organisms from complex mixtures
- Dealing with the ecosystem’s heterogeneity, multiple genomes at varying levels of abundance
- Limiting the reconstruction of chimeras
Assembly strategies
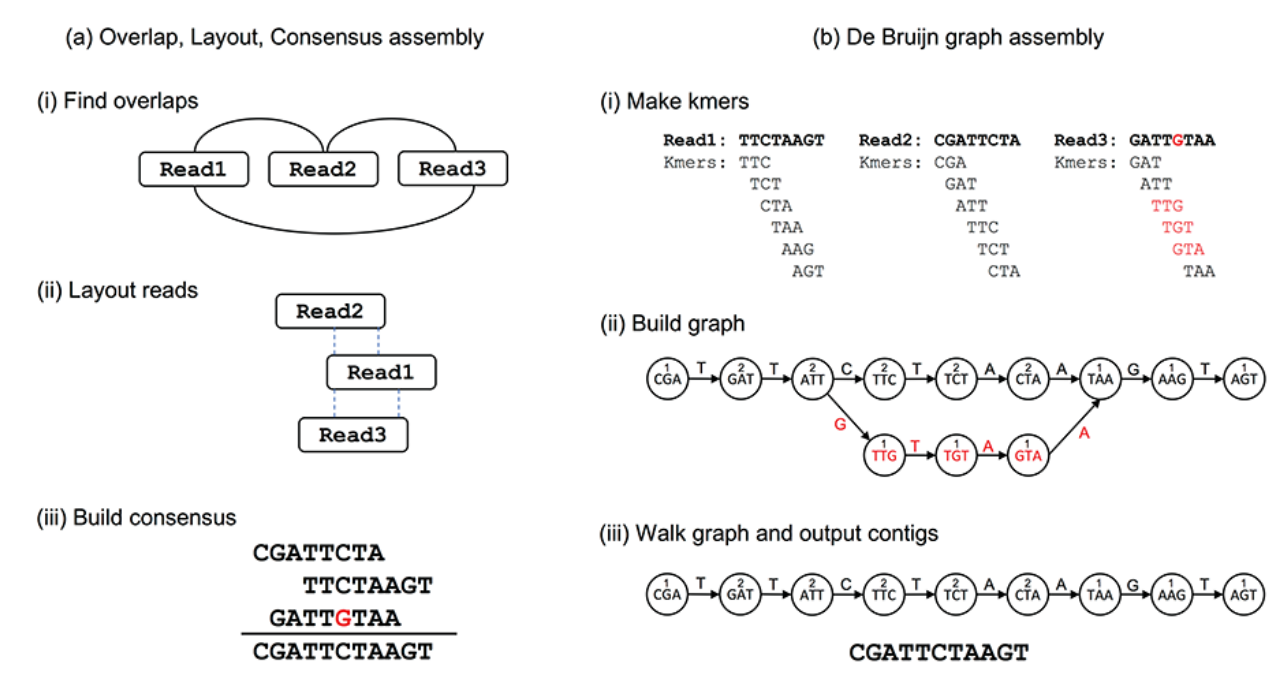
Tools
- Generic tool with a meta option : SPAdes and metaSPAdes [7]
- Tools requiring less memory : MEGAHIT [8]
- The historical tool allowing many parameters : Velvet (and MetaVelvet)
- A (not so) recent benchmark of short reads metagenome assemblers. [9]
- Long read / Hybrid assemblies use different algorithms and strategies and are still a research question.
Assessment of assembly quality
After assembly, we use MetaQUAST [10] to evaluate and compare metagenome assemblies.
What MetaQUAST does :
- De novo metagenomic assembly evaluation
- [Optionally] identify reference genomes from the content of the assembly
- Reference-based evaluation
- Filtering so-called misassemblies based on read mapping
- Report and visulaisation
De novo metrics
Evaluation of the assembly based on:
- Number of contigs greater than a given threshold (0, 1kb, …)
- Total / thresholded assembly size
- Largest contig size
- N50 : the sequence length of the shortest contig at 50% of the total assembly length, equivalent to a median of contig lengths. (N75 idem, for 75%)
- L50 : the number of contigs at 50% of the total assembly length. (L75 idem, for 75%)
Reference-based metrics
- Metrics based on the comparison with reference genomes.
- Reference genomes are given by the user or automatically constitued by MetaQuast based on comparison of rRNA genes content of the assembly and a reference database (Silva).
- Complete genomes are then automatically downloaded.
Reference-based metrics
For each given reference genome, based on an alignement of all contigs on it :
- Duplication rate
- Percent genome complete
- NGA50 : equivalent of N50 but with the aligned block of the contigs
- “Misassemblies” : breakpoint of alignement in a contigs.
- An individual Quast report and an alignement visualisation.
Counts on assembly
- We used only a portion of the reads for assembly.
- For further analyses it is necessary to map all the reads on the contigs to obtain counts per features (genes/contigs…)
- We will use BWA [11]
Switch to TP 5
Binning
Objectives
Binning is a good compromise when the assembly of whole genomes is not feasible.
Similar contigs are grouped together.
Approach
- MetaBAT [14] is a tool for reconstructing genomes from complex microbial communities.
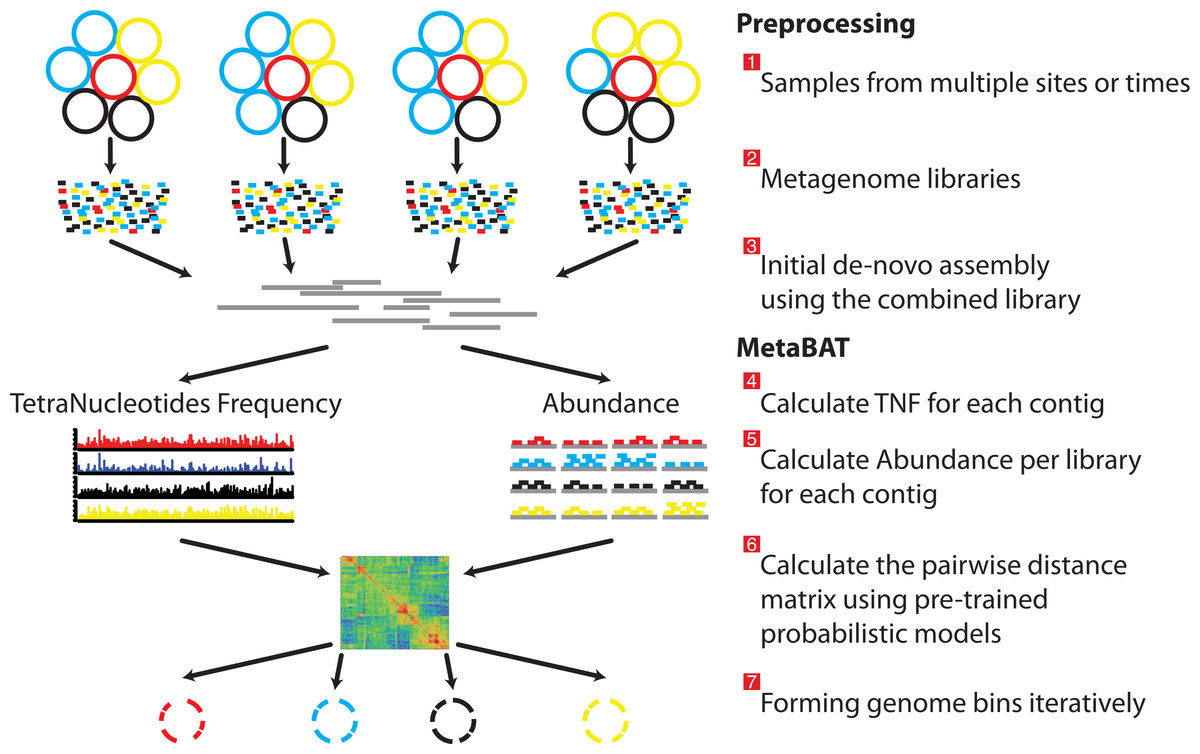
Bins evaluation
For the evaluation of bins, we will use completeness and contamination estimated by CheckM [15]
- Use of collocated sets of genes that are ubiquitous and single-copy within a phylogenetic lineage.
- Among a set of tools in CheckM we will use the
checkm2 predictworkflow which only mandatory requires a directory of genome bins.
Switch to TP 6
Annotation
Objective
- Syntaxic annotation (gene prediction)
- Functionnal annotation (function prediction for protein coding genes)
- Prokka [16] is a is a software tool to annotate bacterial, archaeal and viral genomes (quite) quickly and produce standards-compliant output files.
- Prokka automatically annotate a complete bacterial genome in ~15mn.
- Prokka will not replace expert annotation but gives you an homogenoeus procedure for annotation of conserved genes familly
Genes prediction (1)
- Gene prediction in complete prokaryotic genomes isn’t as such a problem.
- Most errors / difference in start position
- Efficient gene predictors are available (bactgeneSHOW, prodigal, glimmerHMM,…).
- Most of them uses HMM (Hidden Markov Models) models to predict the gene structure
Genes prediction (2)
- Gene prediction on metagenomes is difficult due to :
- assembly fragmentation
- assembly errors, frameshift, chimeras,…
- different species in the same sample that could/should lead to use different models
- a mix of viral, bacterial and eukaryotic sequences.
- Prodigal (with the
-metaparameter) and fraggenescan have good enough results on metagenomic contigs. - Caution to partial genes in the following analysis !
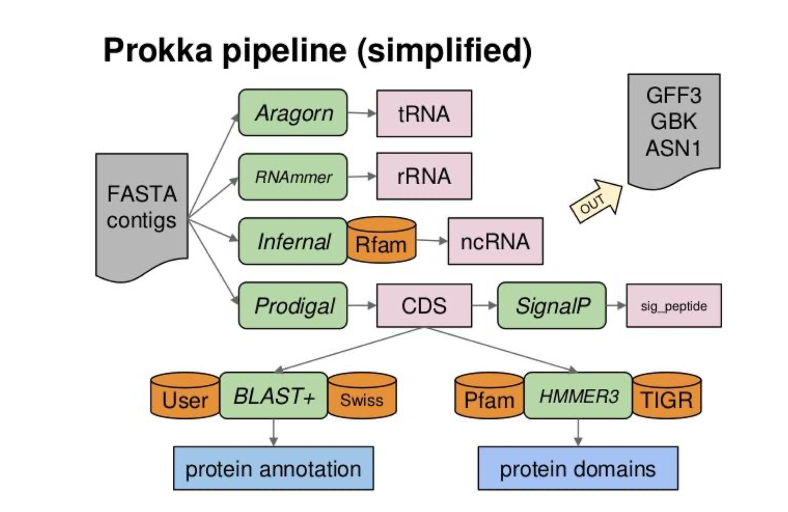
Prokka pipeline
- Coding gene prediction with Prodigal [17]
- tRna; rRna gene prediction with Aragorn, Barnap, RNAmmer (optionnal)
- Functionnal annotation based on similarity search with a threshold (1e-6) and hierarchically against :
- [Optionnal] a given proteome (
--proteinsparameter) - ISFinder for transposases, not entire IS
- NCBI Bacterial Antimicrobial Resistance Reference Gene Database for Antimicrobial Resistance Genes.
- UniprotKB/Swissprot curated proteins with evidence that (i) from Bacteria (or Archaea or Viruses); (ii) not be “Fragment” entries; and (iii) have an evidence level (“PE”) of 2 or lower, which corresponds to experimental mRNA or proteomics evidence.
- Domain and motifs (hmmsearch) :
- [Optionnal] a given proteome (
Prokka pipeline
EggNogg Mapper
- eggNOG-mapper [18] is a tool for fast functional annotation of novel sequences. It uses precomputed orthologous groups and phylogenies from the eggNOG database to transfer functional information from fine-grained orthologs only.
- eggNog database :
- 5090 organisms, 2502 viruses.
- 4.4 M orthologous groups annotated with COG category, Gene Ontolgy, EC number, Kegg orthologs and pathways, CAZy, PFAMs
EggNogg Mapper workflow
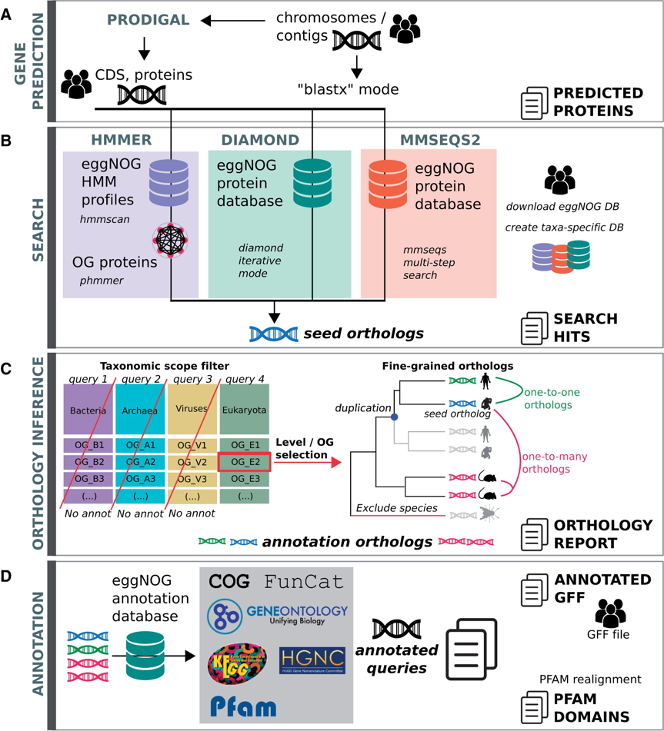
- eggNOG uses hmmsearch to search against HMM eggNOG database OR Diamond / MMSeqs2 to search against eggNOG protein database
- Refine first hit using a list of precomputed orthologs.
- Assign one ortholog
- Functionnal annotation transfer using this ortholog
Other options for functionnal annotation
- Diamond [19] is a sequence aligner for protein (equivalent blastp) and translated DNA (equivalent tblastx) searches, designed for high performance analysis of big sequence data. Diamond is 100x to 20,000x the speed of BLAST.
- Diamond could be used to query against any given databank.
- ghostKOALA [20, Online]
- KOALA (KEGG Orthology And Links Annotation) is KEGG’s internal annotation tool for K number assignment.
Other options for functionnal annotation (2)
- cd-hit [21] is a software for clustering protein sequences. It can be used to downsize the number of lines in the in the gene count matrix.
Switch to TP 7
Biostatistics
What to do after bioinformatics
Automatization
Snakemake workflow
We have developed a workflow that allows us to automate all these analyses.
- developed with snakemake
- executable on MIGALE
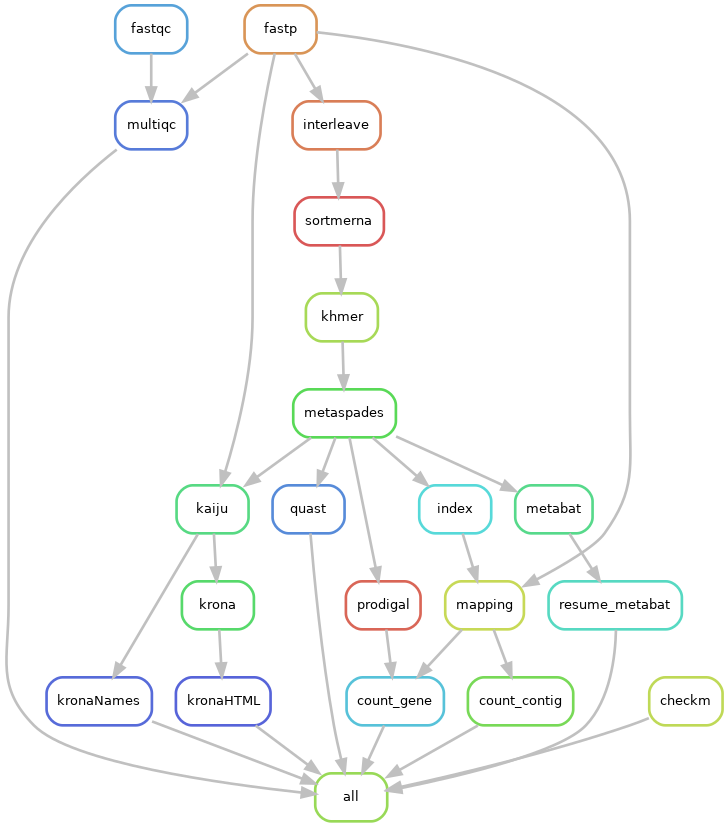
Other tools
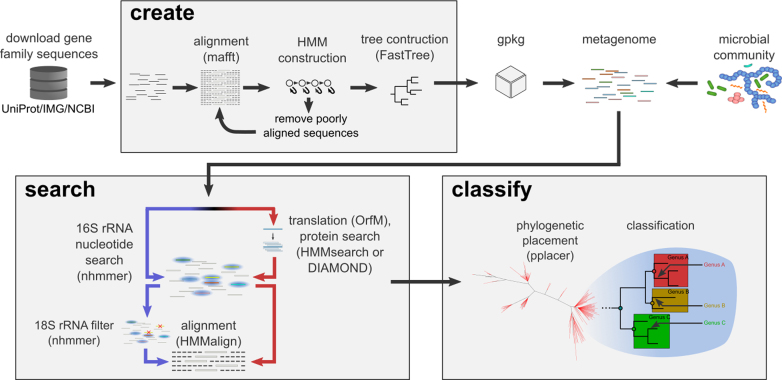
GraftM: rapid community profiles from metagenomes
Anvi’o: integrated multi-omics at scale
- Anvi’o [26] is an open-source, community-driven analysis and visualization platform for microbial ’omics.
- With this tutorial, starting from a metagenomic assembly, you will :
- Process your contigs,
- Profile your metagenomic samples and merge them,
- Visualize your data, identify and/or refine genome bins interactively, and create summaries of your results.
Metagenome atlas
- Metagenome-atlas [27] is an easy-to-use metagenomic pipeline based on
snakemake. - It handles all steps of analysis :
Metagenome atlas
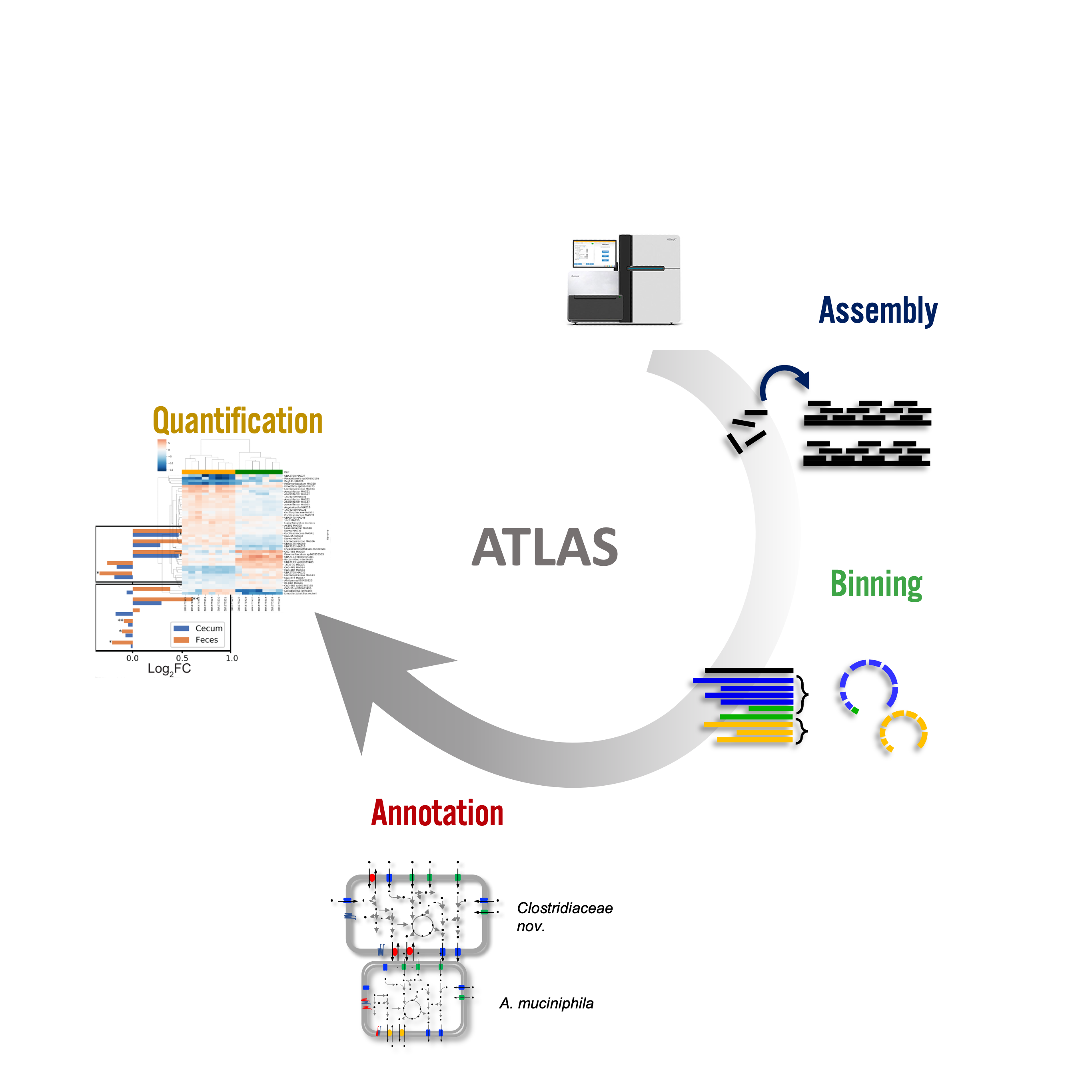
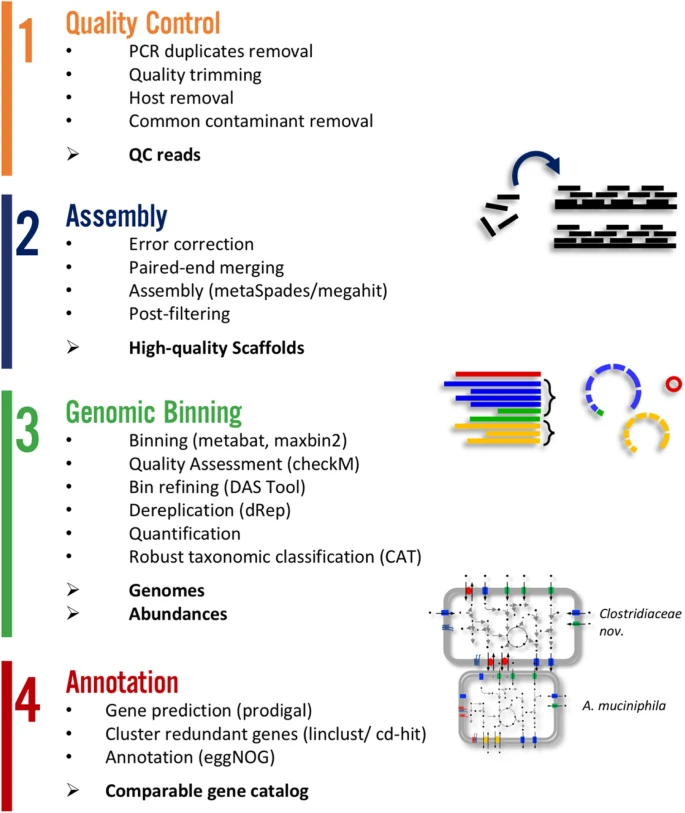
Metagenomics and long reads
- Long read data can be used to improve assembly
- Bottlenecks :
- DNA extraction (?)
- cost of data generation
- sequencing errors
- State of the art pipeline for assembly :
- standalone long read assembly (ex: MetaFLYE [28])
- optional error correction with short reads
Metatranscriptomics
Advances and challenges in metatranscriptomic analysis, 2019 [29]
Take home message
- Shotgun metagenomics is still an ongoing active bioinformatics research field
- Numerous softwares dedicated to assembly, binning, functionnal annotation are actively developed
- Depending on the ecosystem , one can have different approaches :
- mapping on a reference database
- assembly and mapping
- The biological question must determine the analysis
References
1. Escobar-Zepeda A, Vera-Ponce de León A, Sanchez-Flores A. The road to metagenomics: From microbiology to DNA sequencing technologies and bioinformatics. Frontiers in genetics. 2015;6:348.
2. Breitwieser FP, Lu J, Salzberg SL. A review of methods and databases for metagenomic classification and assembly. Briefings in bioinformatics. 2019;20:1125–36.
3. Yang C, Chowdhury D, Zhang Z, Cheung WK, Lu A, Bian Z, et al. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. Computational and Structural Biotechnology Journal. 2021;19:6301–14. doi:https://doi.org/10.1016/j.csbj.2021.11.028.
4. Stoler N, Nekrutenko A. Sequencing error profiles of Illumina sequencing instruments. NAR Genomics and Bioinformatics. 2021;3. doi:10.1093/nargab/lqab019.
5. Wood DE, Salzberg SL. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome biology. 2014;15:1–12.
6. Menzel P, Ng KL, Krogh A. Fast and sensitive taxonomic classification for metagenomics with kaiju. Nature communications. 2016;7:11257.
7. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 2012;19:455–77. doi:10.1089/cmb.2012.0021.
8. Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics. 2015;31:1674–6.
9. Vollmers J, Wiegand S, Kaster A-K. Comparing and evaluating metagenome assembly tools from a microbiologist’s perspective-not only size matters! PloS one. 2017;12:e0169662.
10. Mikheenko A, Saveliev V, Gurevich A. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics. 2015;32:1088–90. doi:10.1093/bioinformatics/btv697.
11. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997. 2013.
12. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
13. Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
14. Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165.
15. Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research. 2015;25:1043–55. doi:10.1101/gr.186072.114.
16. Seemann T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. doi:10.1093/bioinformatics/btu153.
17. Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC bioinformatics. 2010;11:1–11.
18. Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, Mering C von, et al. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Molecular Biology and Evolution. 2017;34:2115–22. doi:10.1093/molbev/msx148.
19. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nature Methods. 2014;12:59–60. doi:10.1038/nmeth.3176.
20. Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. Journal of molecular biology. 2016;428:726–31.
21. Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–2. doi:10.1093/bioinformatics/bts565.
22. Liu Y-X, Qin Y, Chen T, Lu M, Qian X, Guo X, et al. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein & Cell. 2020;12:315–30. doi:10.1007/s13238-020-00724-8.
23. Benoit G, Mariadassou M, Robin S, Schbath S, Peterlongo P, Lemaitre C. SimkaMin: Fast and resource frugal de novo comparative metagenomics. Bioinformatics. 2019. doi:10.1093/bioinformatics/btz685.
24. Steinegger M, Söding J. Clustering huge protein sequence sets in linear time. Nature Communications. 2018;9. doi:10.1038/s41467-018-04964-5.
25. Steinegger M, Mirdita M, Söding J. Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nature Methods. 2019;16:603–6. doi:10.1038/s41592-019-0437-4.
26. Eren AM, Esen OC, Quince C, Vineis JH, Morrison HG, Sogin ML, et al. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ. 2015;3:e1319. doi:10.7717/peerj.1319.
27. Kieser S, Brown J, Zdobnov EM, Trajkovski M, McCue LA. ATLAS: A snakemake workflow for assembly, annotation, and genomic binning of metagenome sequence data. BMC Bioinformatics. 2020;21. doi:10.1186/s12859-020-03585-4.
28. Kolmogorov M, Rayko M, Yuan J, Polevikov E, Pevzner P. metaFlye: Scalable long-read metagenome assembly using repeat graphs. 2019. doi:10.1101/637637.
29. Shakya M, Lo C-C, Chain PS. Advances and challenges in metatranscriptomic analysis. Frontiers in genetics. 2019;10:904.

Module 24 - Métagénomique shotgun