Analyse de données métagénomiques 16S
Module 20

September 11, 2023
Better know us

- Open infrastructure dedicated to life sciences
- Computing resources, tools, databanks…
- Dissemination of expertise in bioinformatics
- Design and development of applications
- Data analysis
Data analysis service
- We are specialized in genomics/metagenomics
- 3 Bioinformaticians and 2 Statisticians
- More than 140 projects since 2016
- 2 types of partnership
- Classical collaboration (we perfom the analyses)
- Accompaniment (we help you do the analysis yourself)


Our expectations

Aim of this training

Meta-omics using next-generation sequencing (NGS)

Meta-omics using next-genertation sequencing (NGS)

Strengths and weaknesses of amplicon analyses?

The gene marker power

Microbial tree of life

The 16S resolution


16S rRNA copy number
Median of the number of 16S rRNA copies in 3,070 bacterial species according to data reported in rrnDB database – 2018

16S rRNA copy variation

[B] The positions of sequence variation within 16S and 23S rRNA are shown along the gene organization of rrn operons. A total of 33 and 77 differences were identified in 16S rRNA and 23S rRNA, respectively.
[C] The number of bases that are different from the conserved sequence are shown for 16S and 23S rRNA for each rrn operon
Fungal ITS
ITS: Internal Transcribed Spacer
Size polymorphism of ITS (from 361 to 1475 bases in UNITE 7.1)
Highly conserved regions of the neighboring of ITS1 and ITS2
Lack of a generalist and abundant ITS databank (several small specialized databanks)
Multiple copies (14 to 1400 copies (mean at 113, median at 80))
FROGS deals very good with ITS [8]
- small and long fragments contrary to many tools

Challenges

Amplification bias

- C and D impact the abundance without adding new sequences
- E and F add new sequences
Sequencing technologies
Sequencing technologies

Sequencing technologies

Illumina technology

Illumina technology

Effect of sequencing technology

Interest of controls

Interest of controls

Illustration
Here, we showed that contaminant OTUs from extraction and amplification steps can represent more than half the total sequence yield in sequencing runs, and lead to unreliable results when characterizing tick microbial communities. We thus strongly advise the routine use of negative controls in tick microbiota studies, and more generally in studies involving low biomass samples

Synthetis of biases

Synthesis
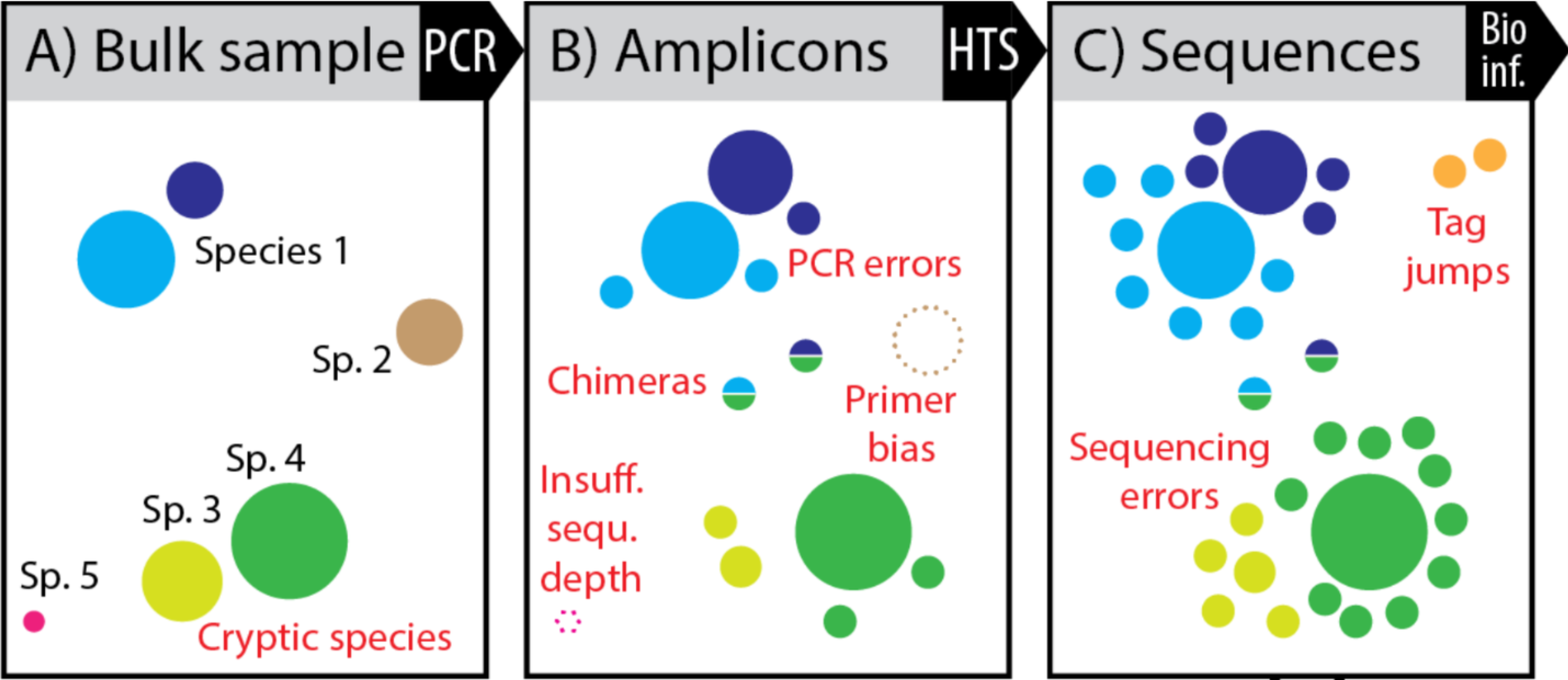
A pile of pipelines


Benchmarking

Compositions at the phylum level for Human gut and, using a range of different methods (separate subpanels within each group).
Benchmarking
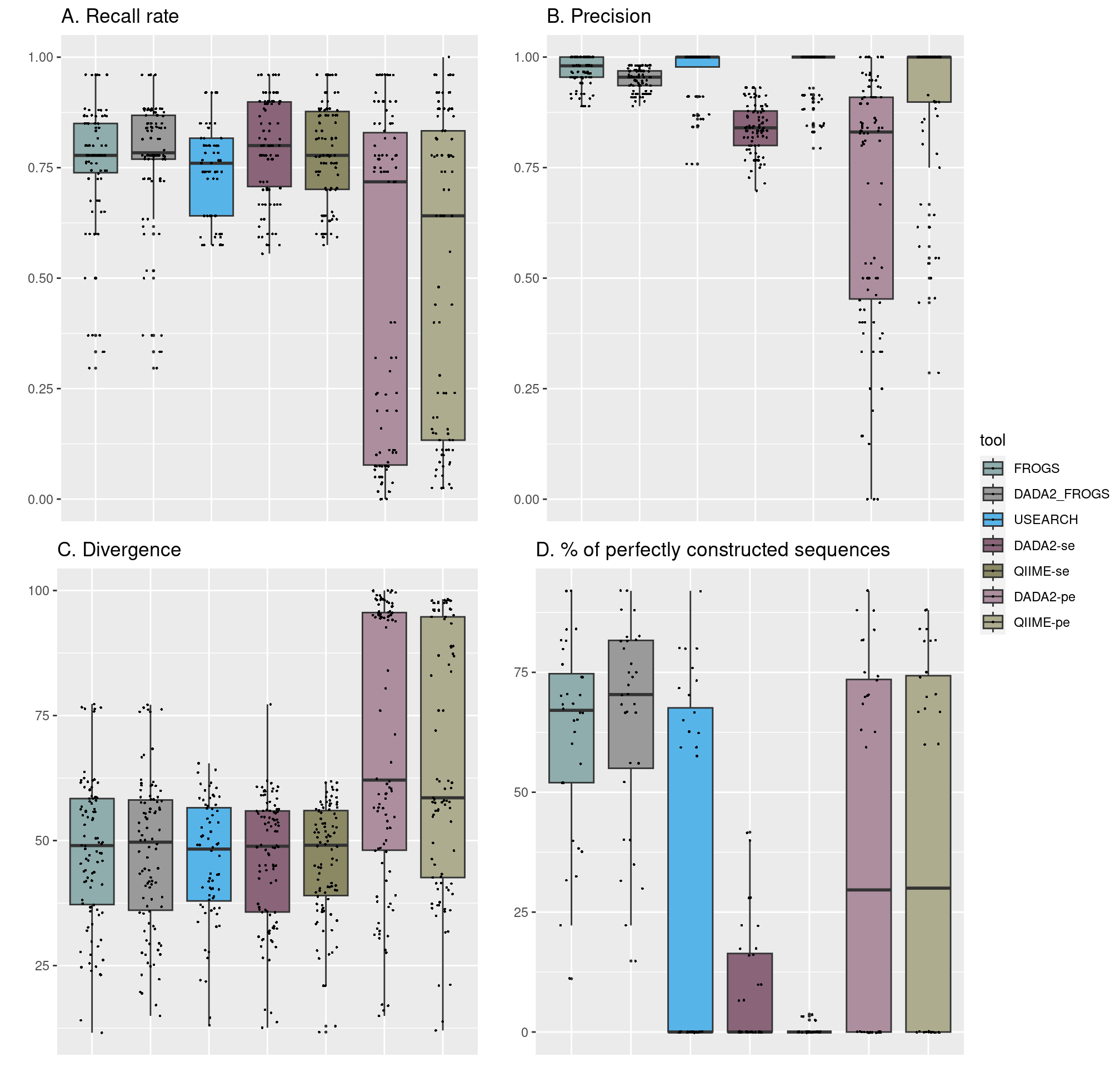
Quality parameters obtained with the seven bioinformatics pipelines. A) Recall rate (TP/(TP+FN)) reflects the capacity of the tools to detect expected species. B) Precision (TP/(TP+FP)) shows the fraction of relevant species among the retrieved species. C) Divergence rate is the Bray-Curtis distance between expected and observed species abundance. D. Percentage of perfectly reconstructed sequences is the fraction of predicted sequences with 100% of identity with the expected ones.
Summary

