Analyse de données NGS sous Galaxy
Module 8 bis

MaIAGE
PROSE & MaIAGE
April 23, 2024
Better know us

- Open infrastructure dedicated to life sciences
- Computing resources, tools, databanks…
- Dissemination of expertise in bioinformatics
- Design and development of applications
- Data analysis
The truth about bioinformatics
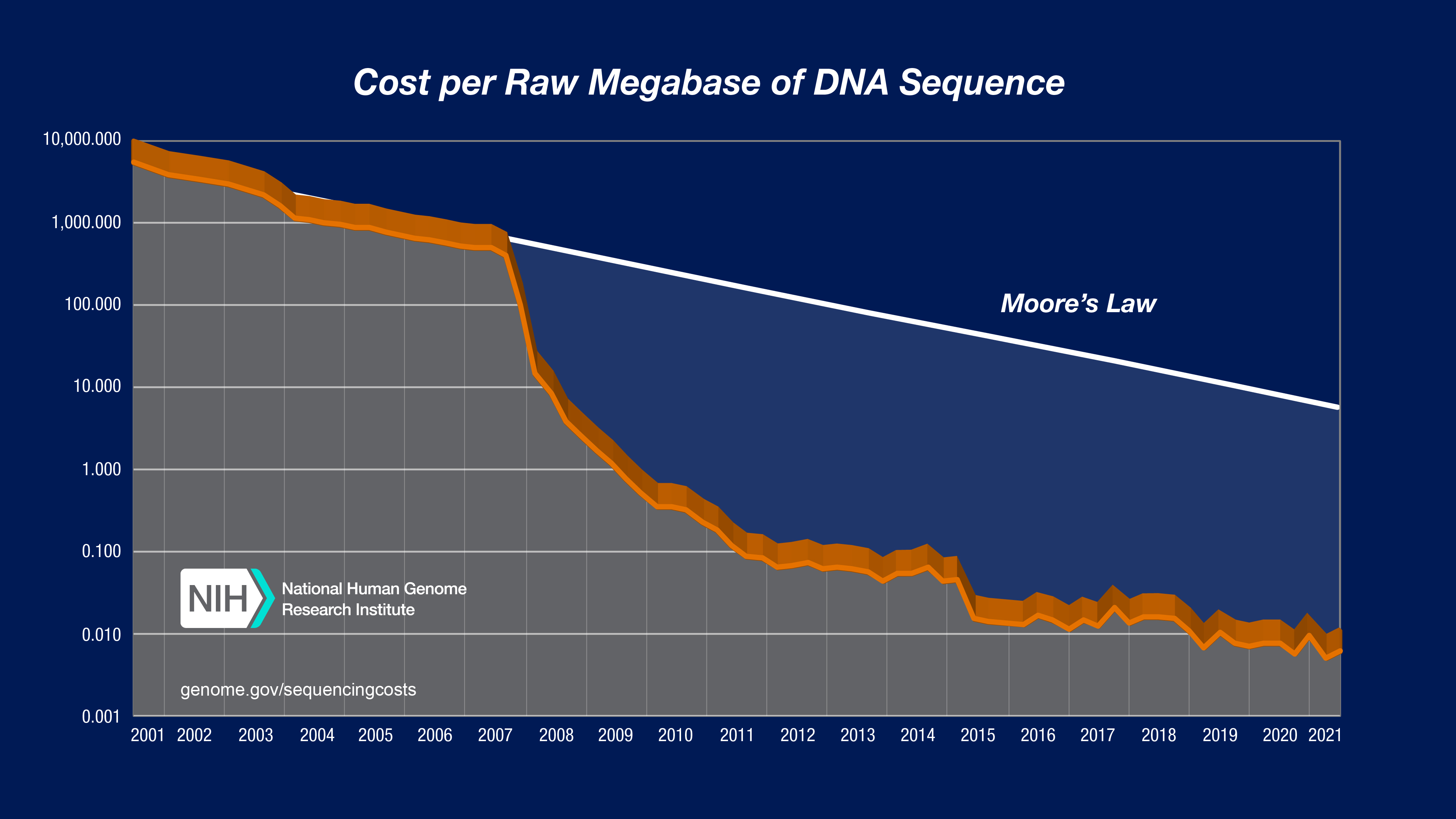
Sequencing Cost per Megabase
Prepare genomic DNA samples
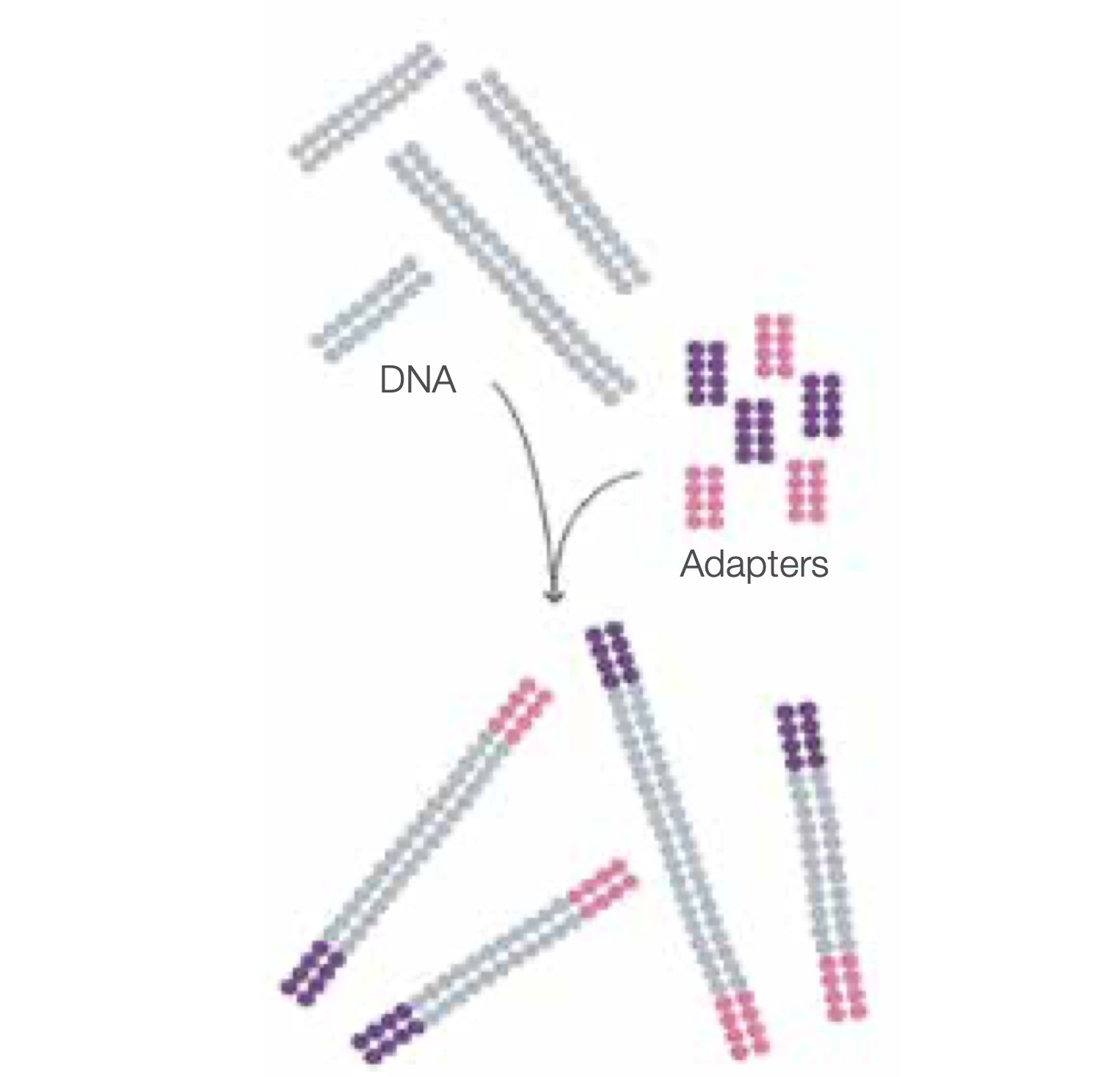
Randomly fragment genomic DNA and ligate adapters to both ends of the fragments
Attach DNA to Flow Cell Surface
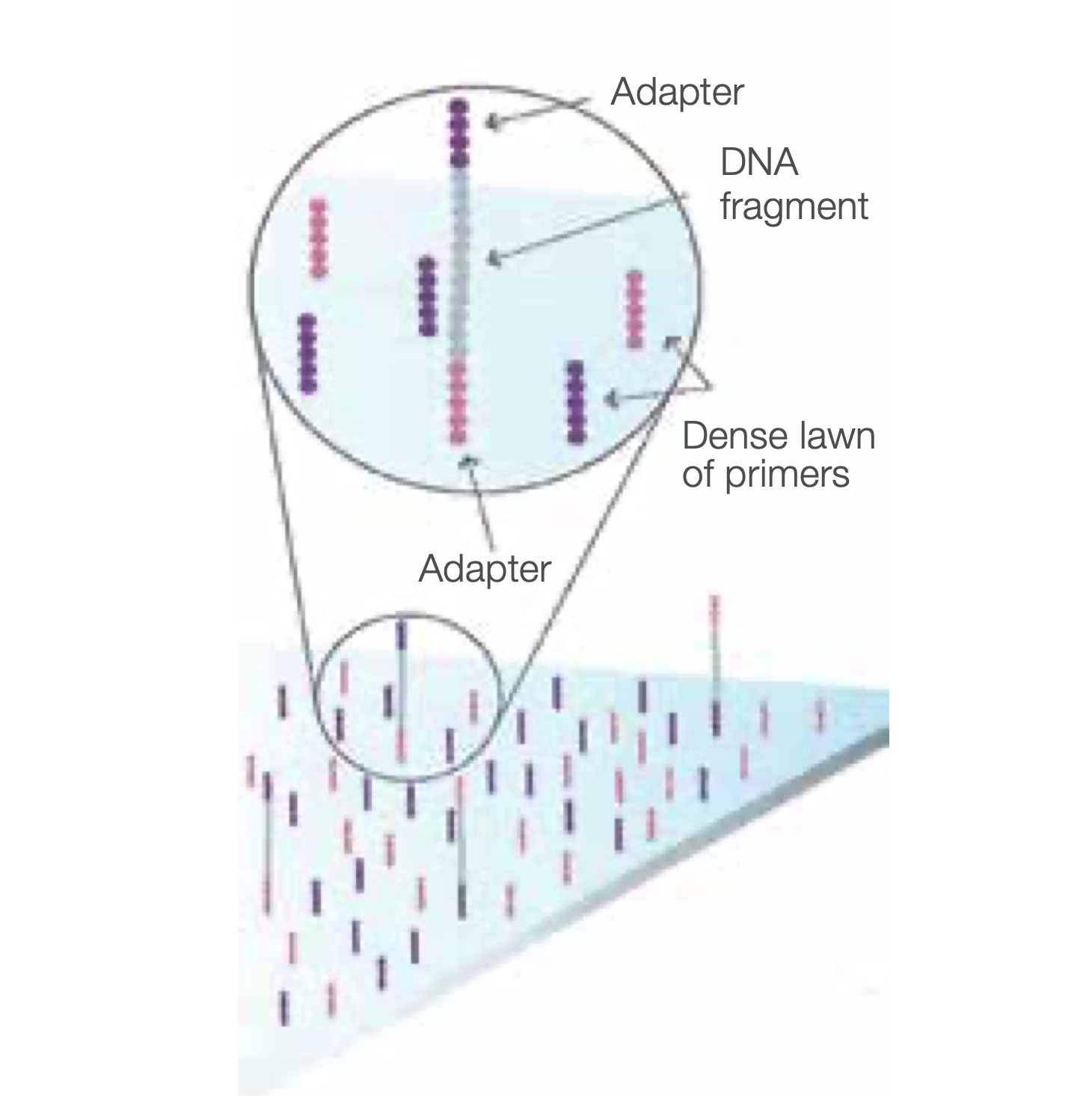
Bind single-stranded fragments randomly to the inside surface of the flow cell channels.
Bridge Amplification
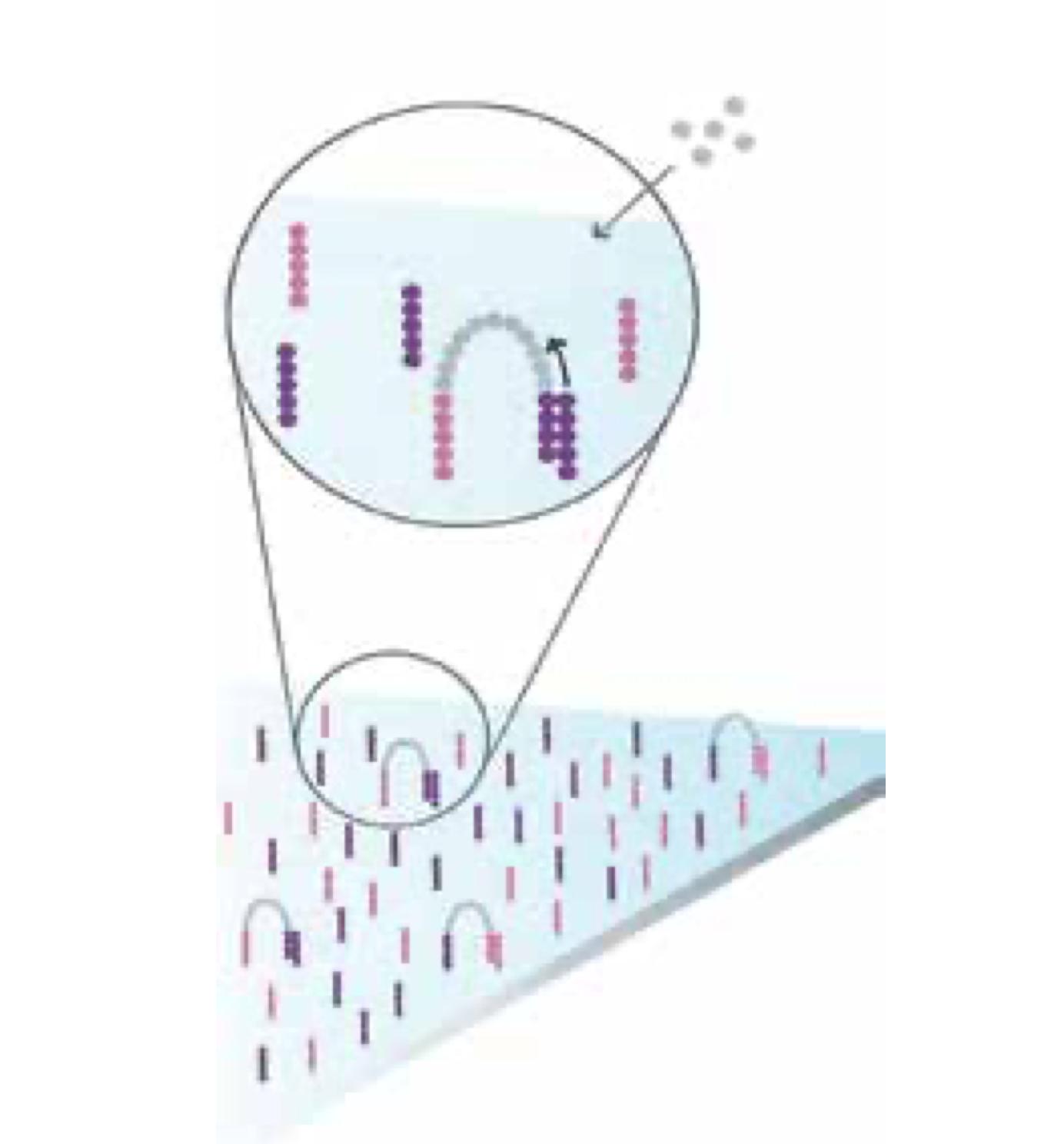
Add unlabelled nucleotides and enzyme to initiate solid-phase bridge amplification.
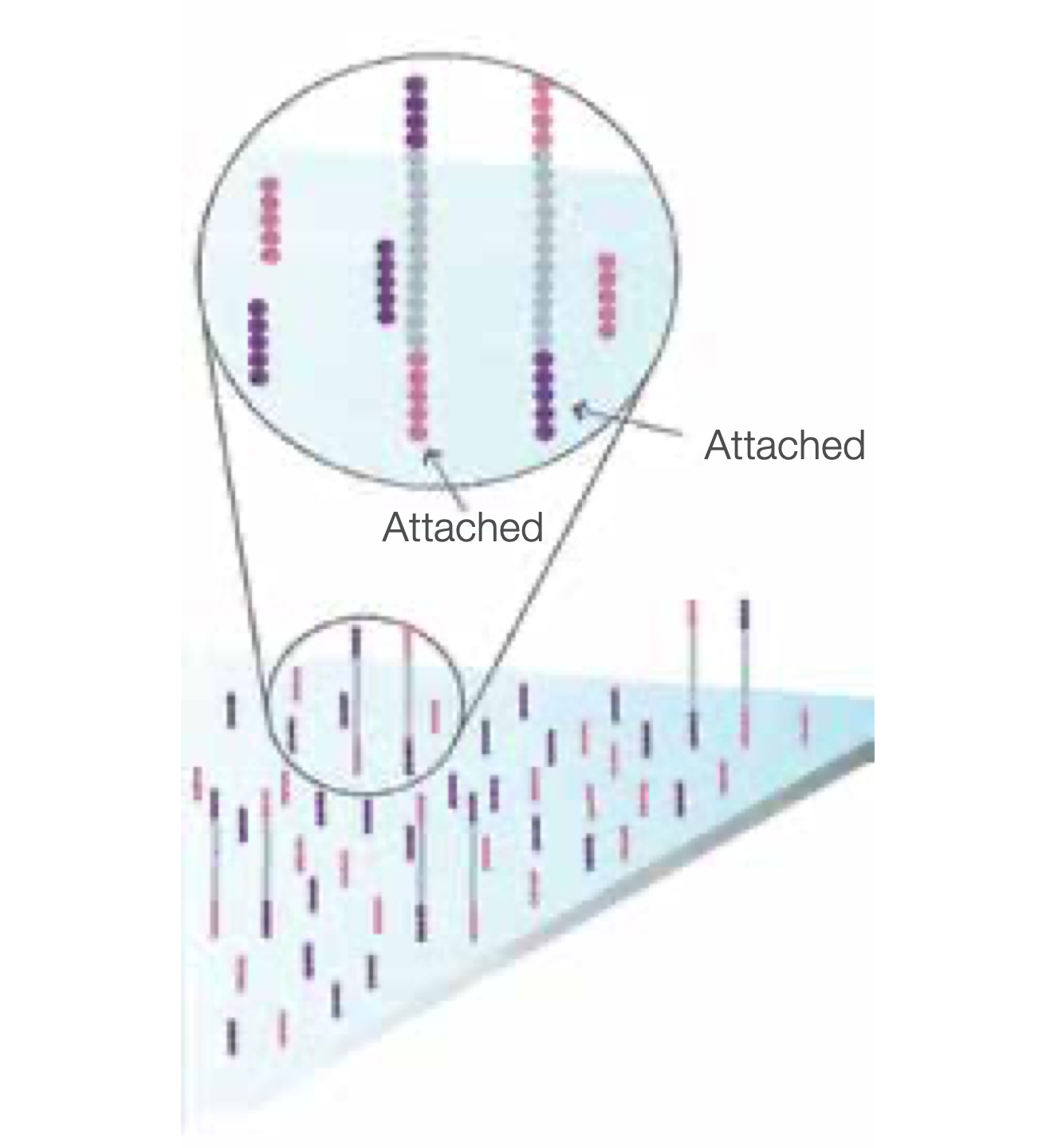
Fragments Become Double Stranded
The enzyme incorporates nucleotides to build double-stranded bridges on the solid-phase substrate.
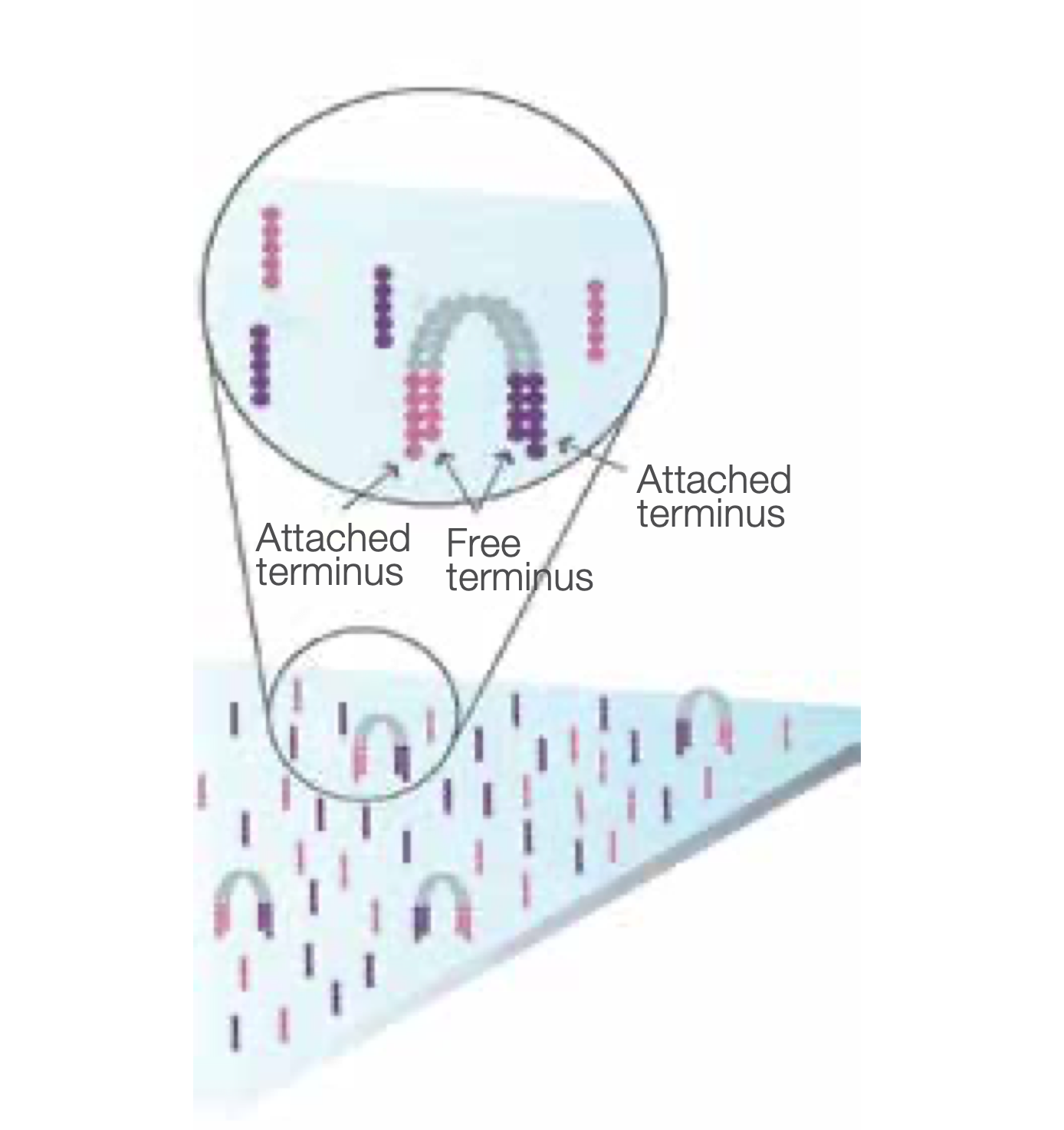
Denature the Double-Stranded Molecule
Denaturation leaves single-stranded templates anchored to the substrate.
Complete Amplification
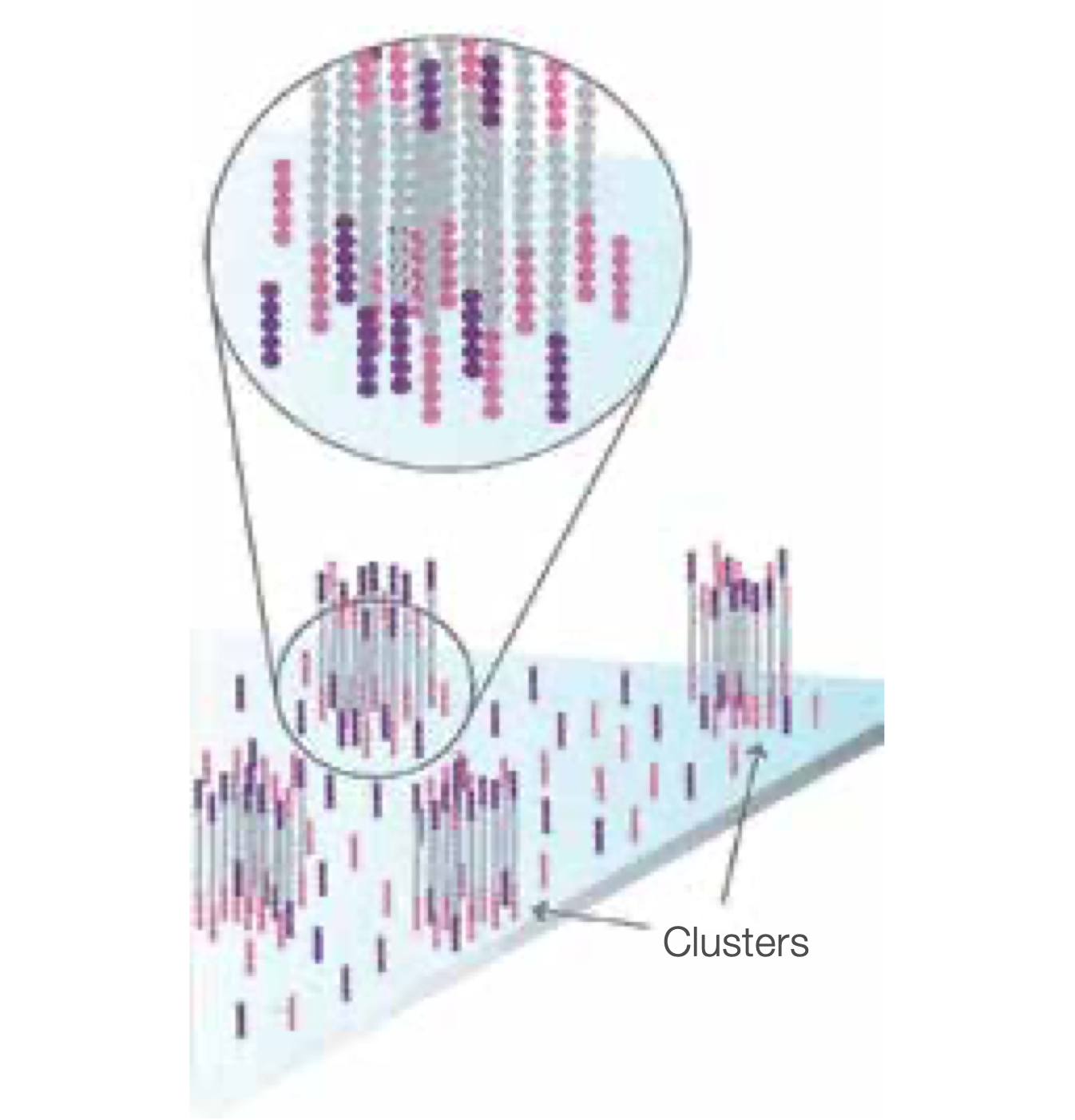
Several millions dense clusters of double-stranded DNA are grated in in channel of the flow cell.
Determine First Base
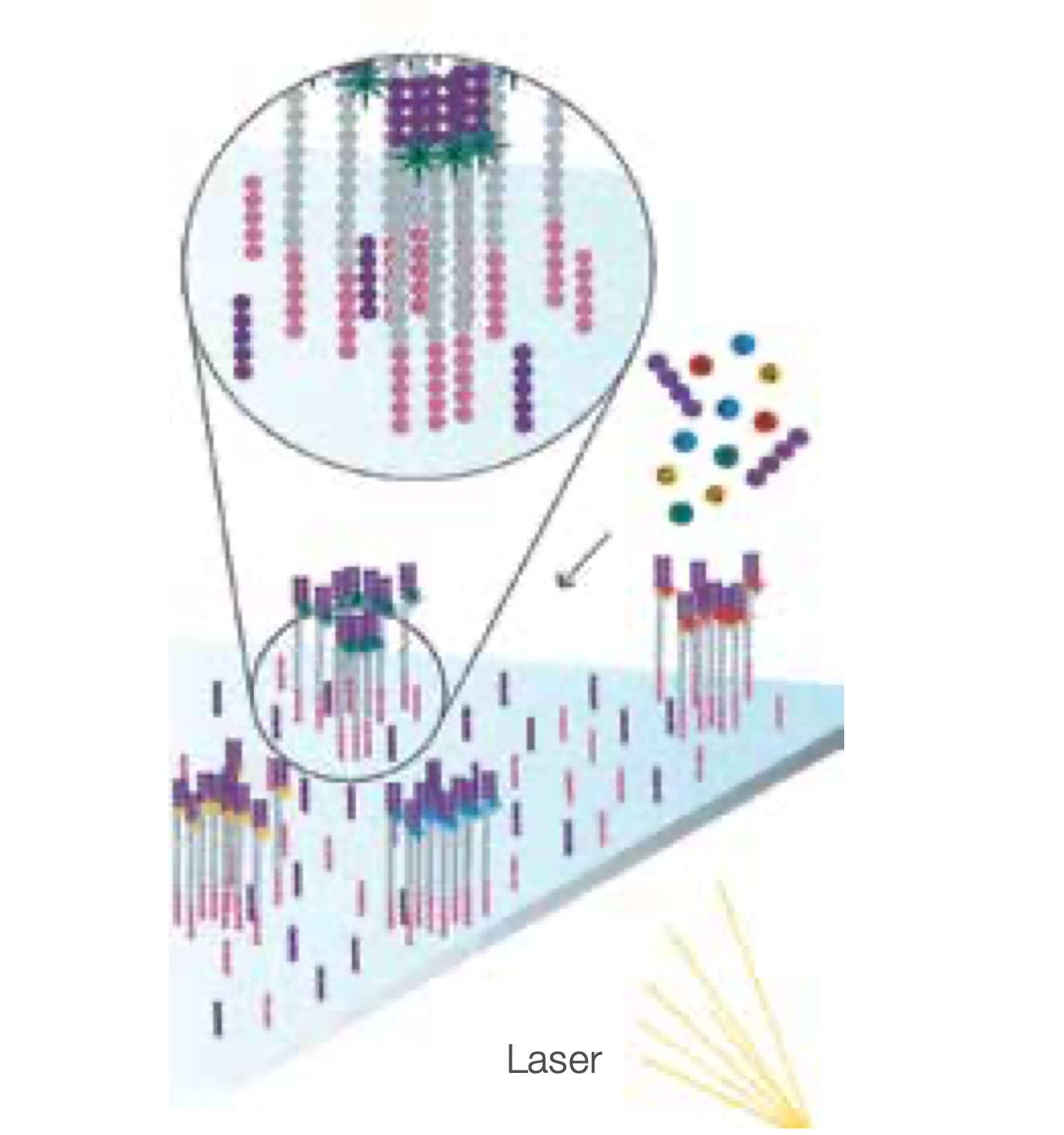
The first sequencing cycle begins by adding four labelled reversible terminators, primers, and DNA polymerase.
Image First Base

After laser excitation, the emitted fluorescence from each cluster is captured and the first base is identified.
The blocked 3’ terminus and florphore are removed,flow cell washed, leaving the terminator free for a second cycle.
Determine Second Base
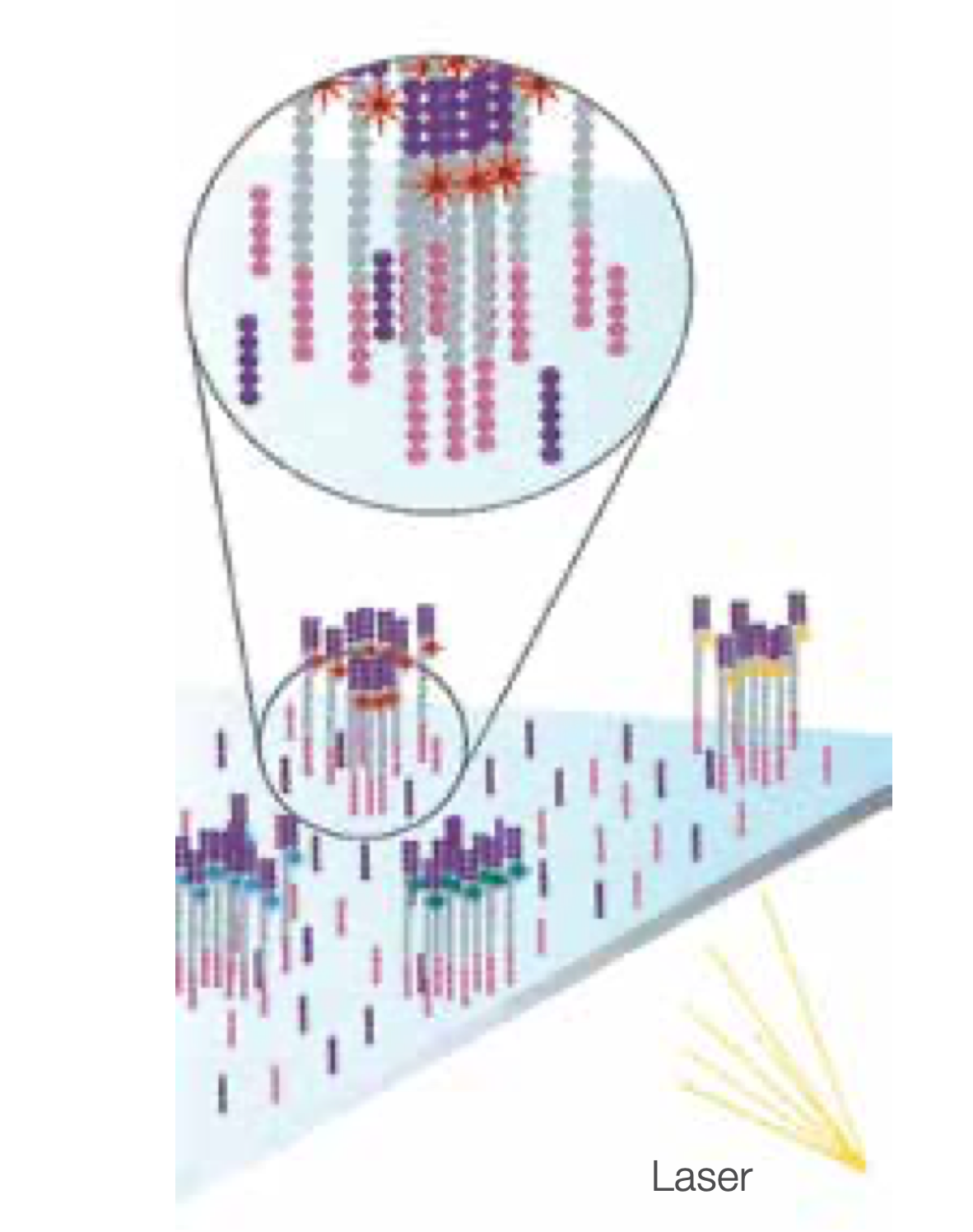
The next cycle repeats the incorporation of four labelled reversible terminators, primers, and DNA polymerase.
Image Second Chemistry Cycle
After laser excitation, the image is captured as before, and the identity of the second base is recorded.
Sequencing Over Multiple Chemistry Cycles
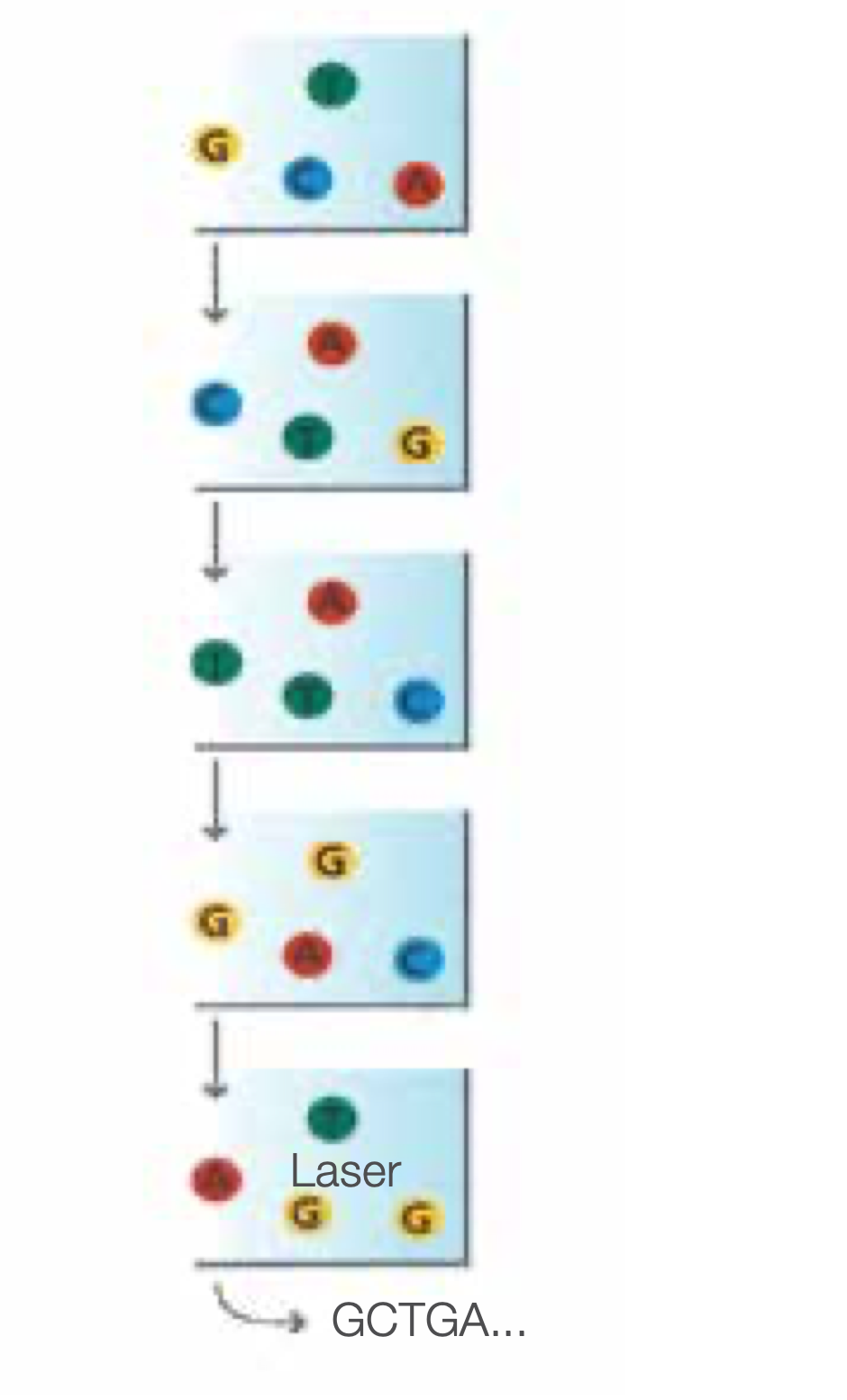
The sequencing cycles are repeated to determine the sequence of bases in a fragment, one base at a time.
Millions of clusters are processed in parallel, allowing high-throughput sequencing.
Sequencing - Vocabulary
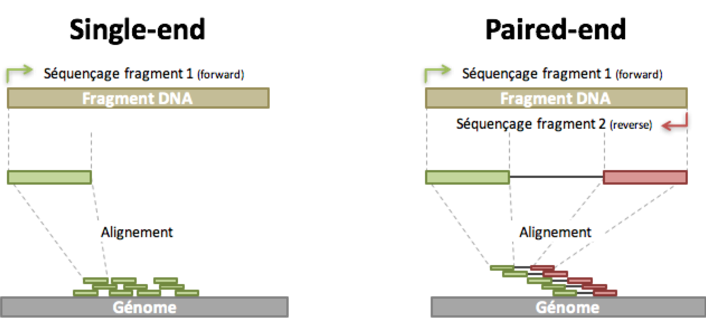
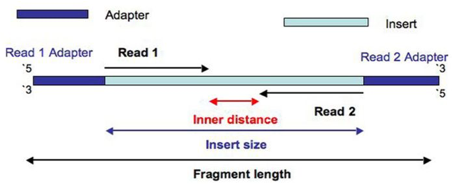
- Read: piece of sequenced DNA
- DNA fragment: 1 or more reads depending on whether the sequencing is single- or paired-end
- Insert: Fragment size
- Depth: \(\frac{N * L}{G}\)
\(N =\) number of reads
\(L =\) reads size
\(G =\) genome size - Coverage: % of genome covered

PacBio
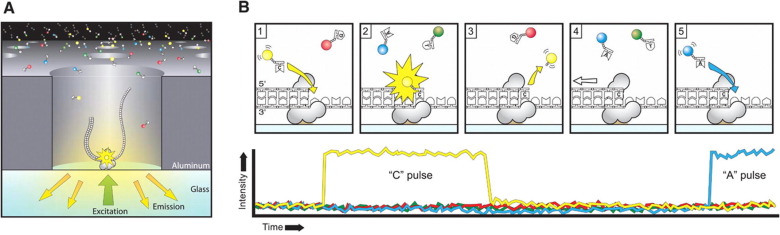
A polymerase is immobilized at the bottom of a sequencing unit called zero-mode waveguide (ZMW) .Four fluorescent-labelled nucleotides, which generate distinct emission spectrums, are added to the SMRT cell. As a base is held by the polymerase, a light pulse is produced that identifies the base. The replication processes in all ZMWs of a SMRT cell are recorded by a “movie” of light pulses, and the pulses corresponding to each ZMW can be interpreted to be a sequence of bases.
Oxford Nanopore

Oxford Nanopore
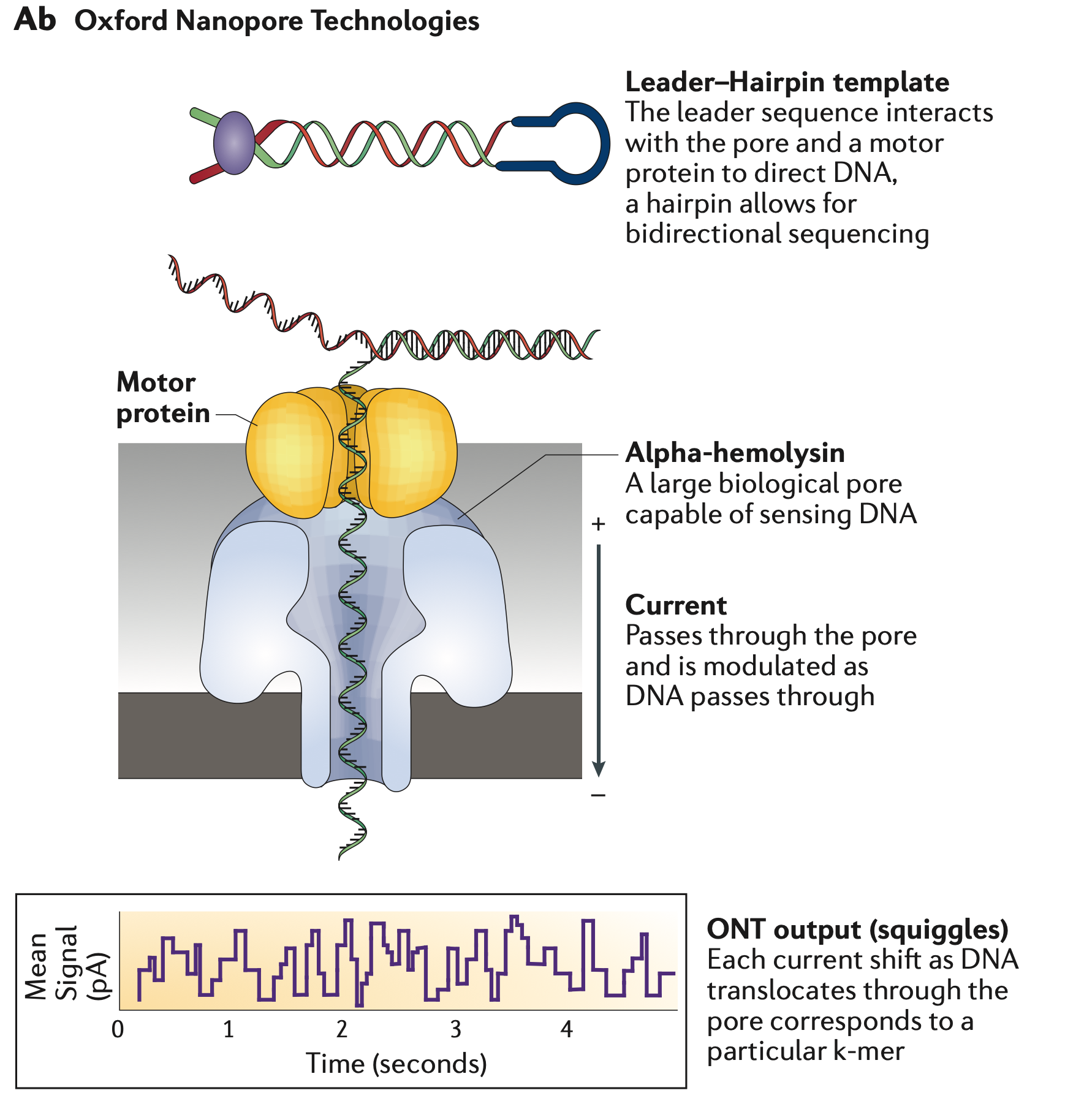
MinION, GridION, PromethION

Sequencing on The ISS

An other view on sequencing technologies (probably out of date)
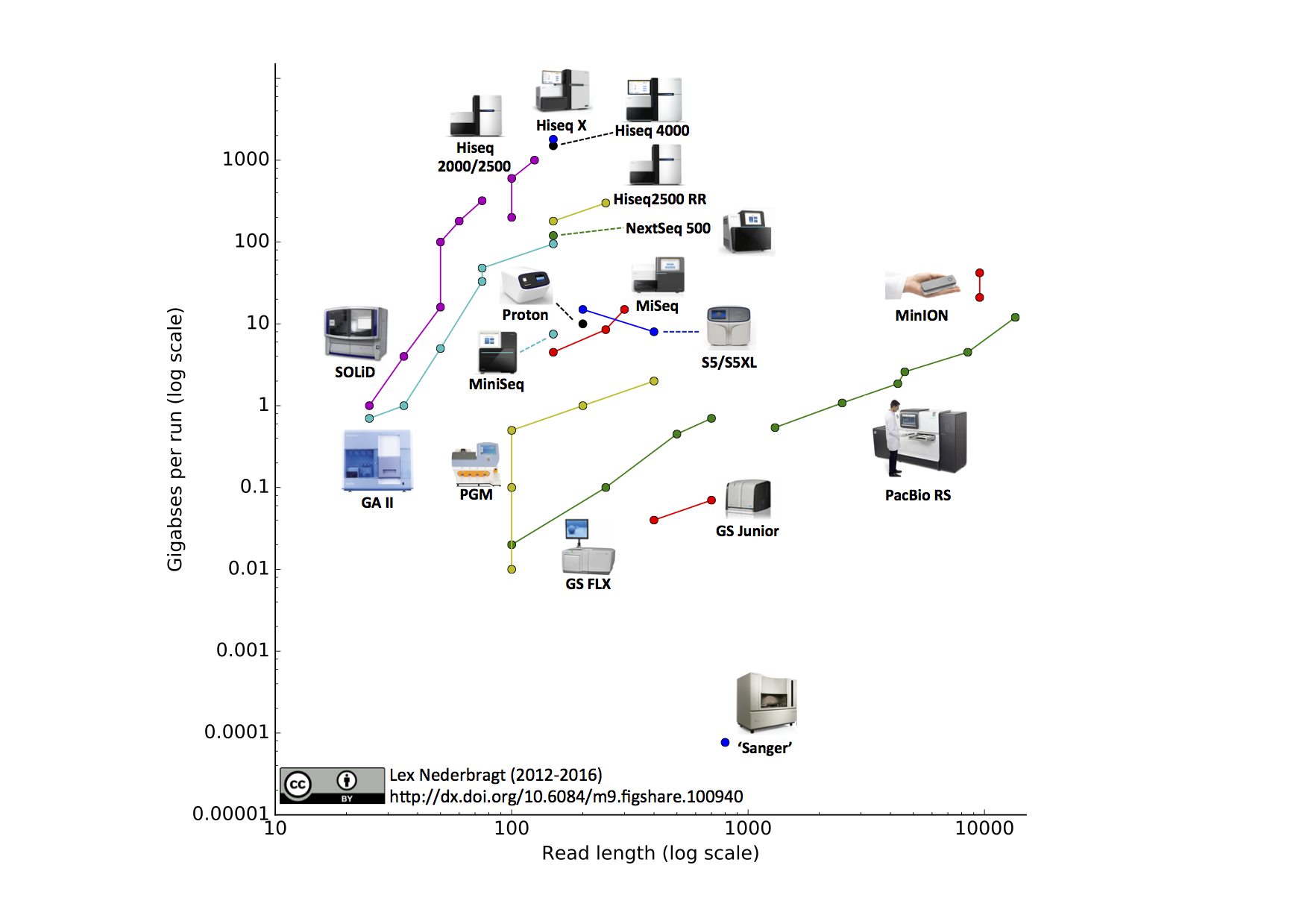
Global Summary (probably out of date)
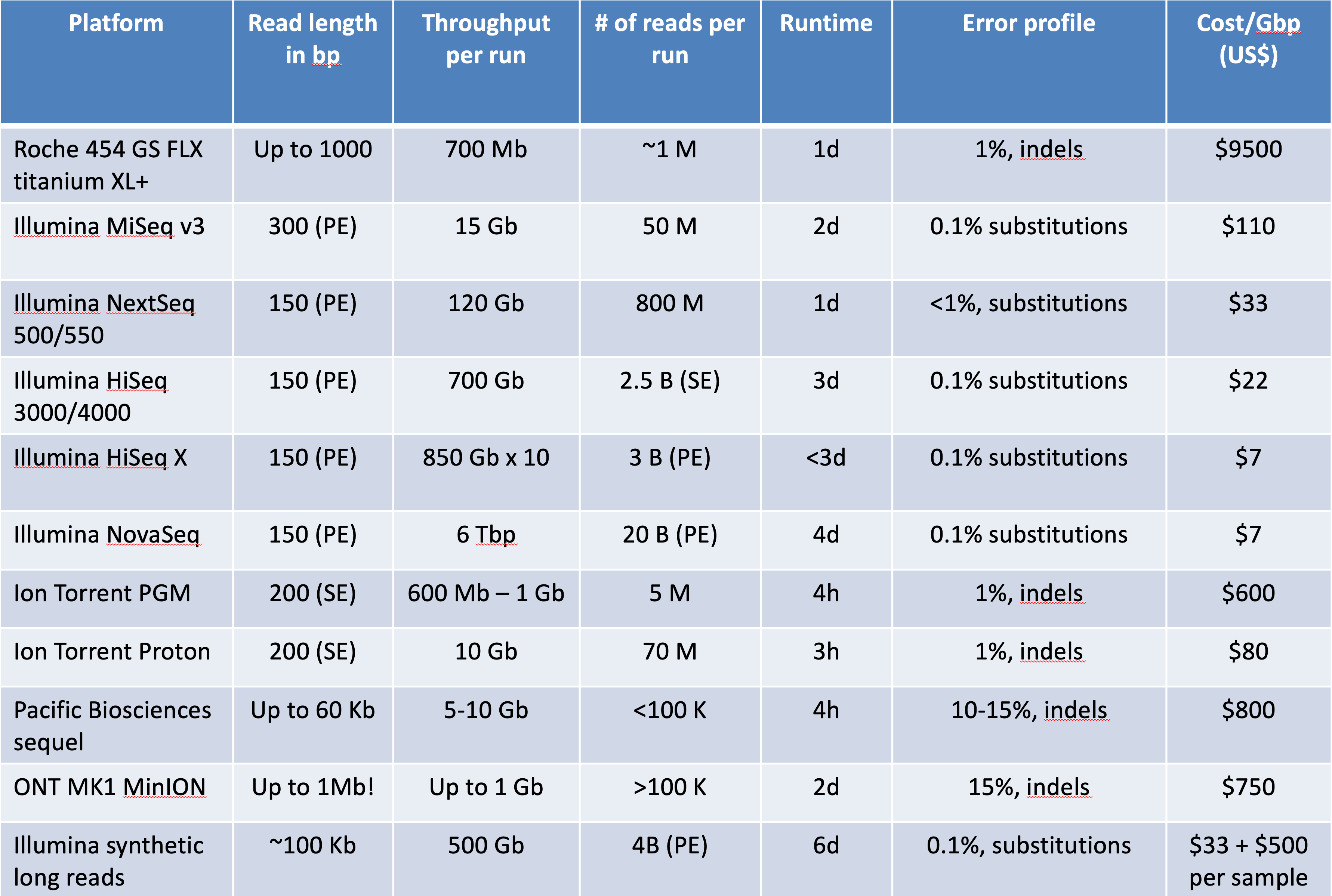
An interesting review [1]
Nature review : Milestones in Genomic Sequencing
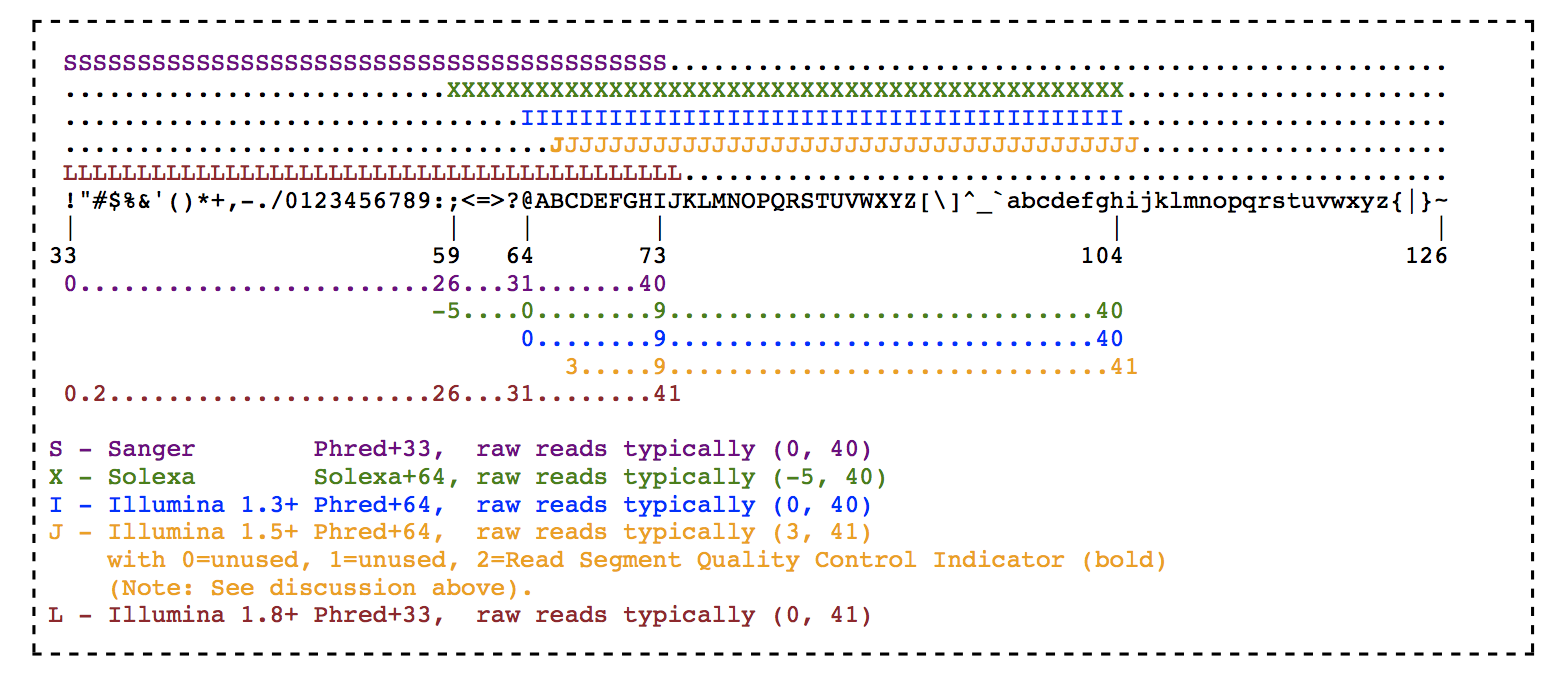
FASTQ quality encoding specificities
There was a time when instrumentation makers could not decide at what character to start the scale. The current standard shown above is the so-called Sanger (+33) format where the ASCII codes are shifted by 33. There is the so-called +64 format that starts close to where the other scale ends.
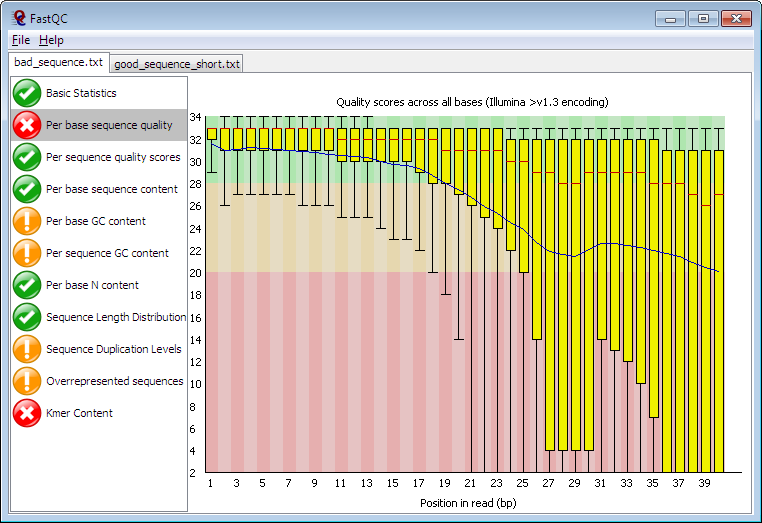
Quality control for FASTQ files : FastQC [2]
- QC for (Illumina) FastQ files
- Command line fastqc or graphical interface
- Complete HTML report to spot problem originating from sequencer, library preparation, contamination
- Summary graphs and tables to quickly assess your data
Tools
- Simple & fast :
Sickle[3] (quality) - dedicated to adapters
cutadapt[4] (adpater removal) - Ultra-configurable :
Trimmomatic - All in one & ultra-fast :
fastp[5]
Assembly : principles
Similar to a puzzle :
- millions of pieces
- without the original image
- with pieces in both sense
- the pieces do not necessarily fit together (sequencing errors)
- parts of the puzzle are missing (cover + sequencing bias)

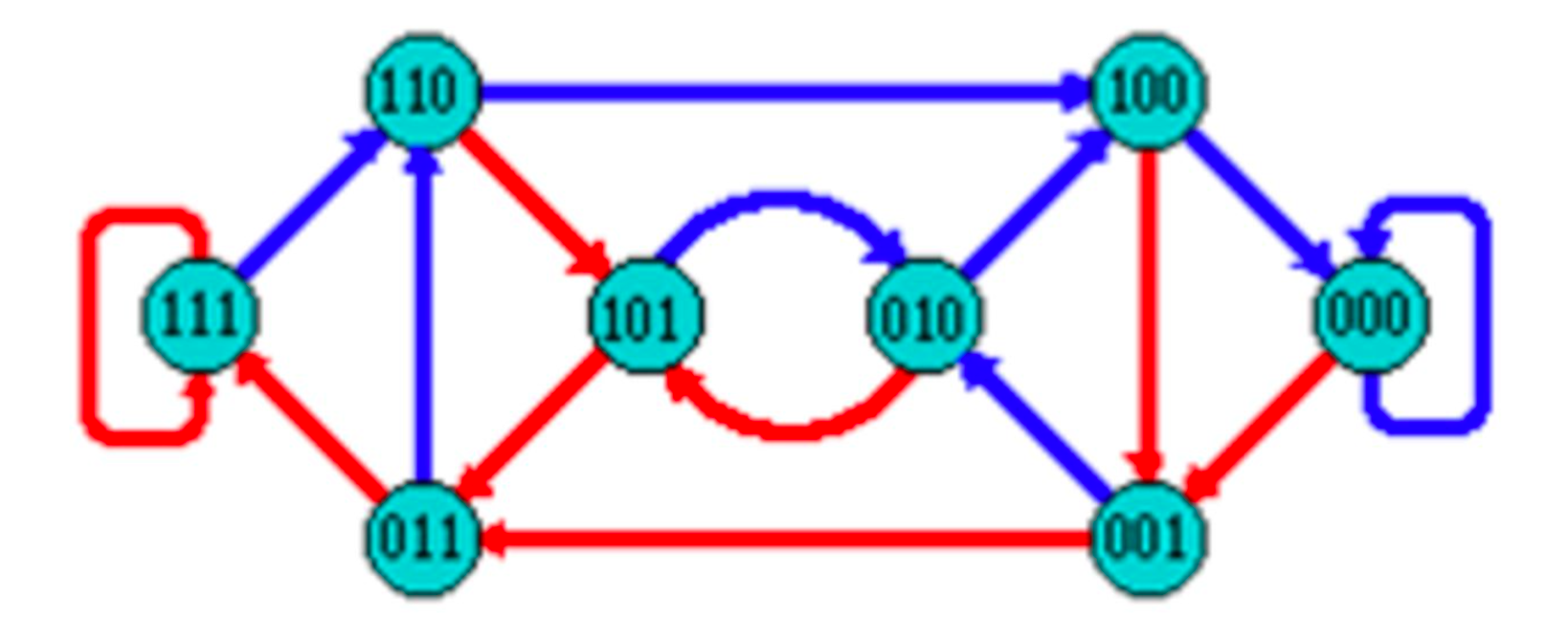
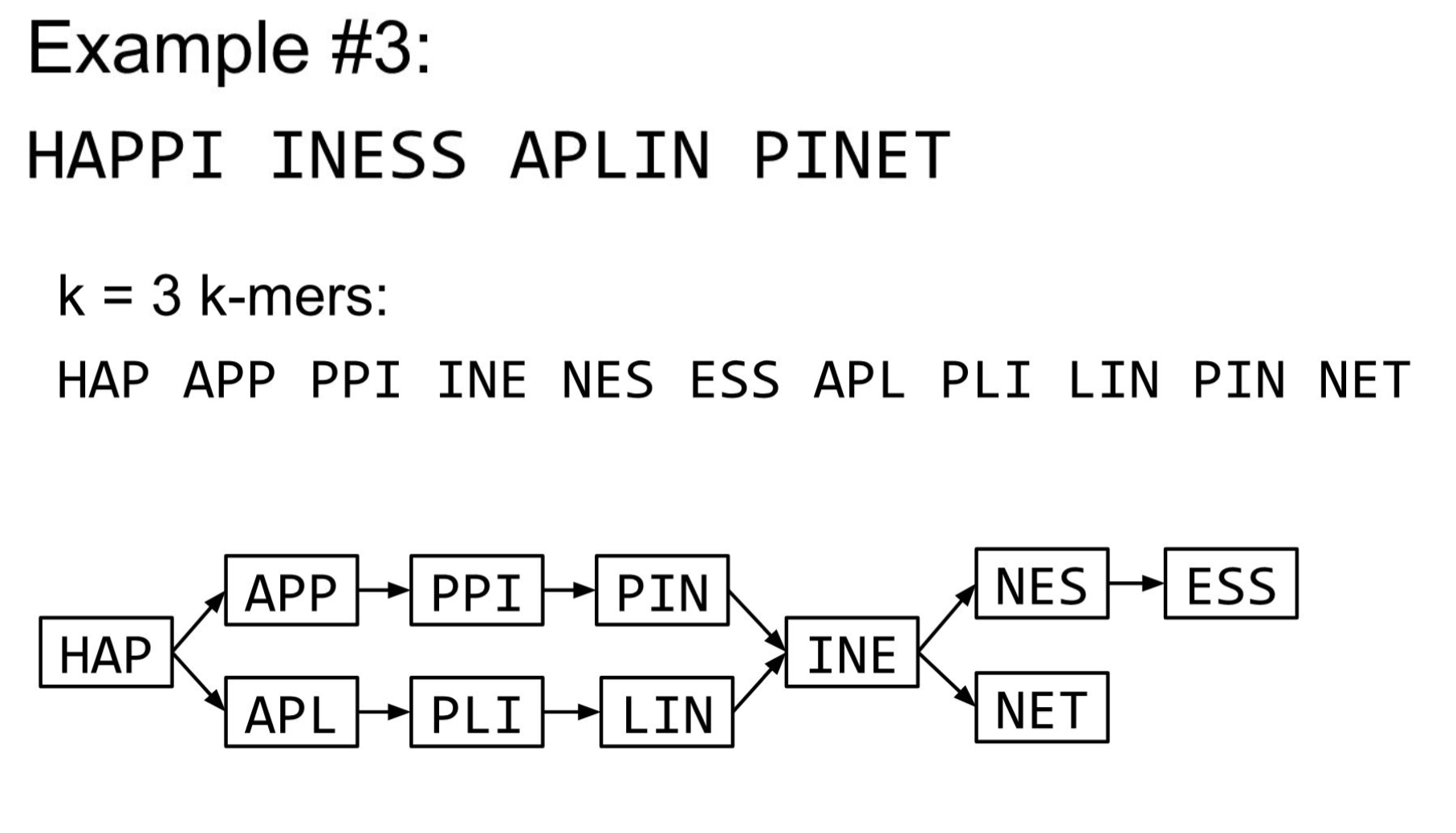
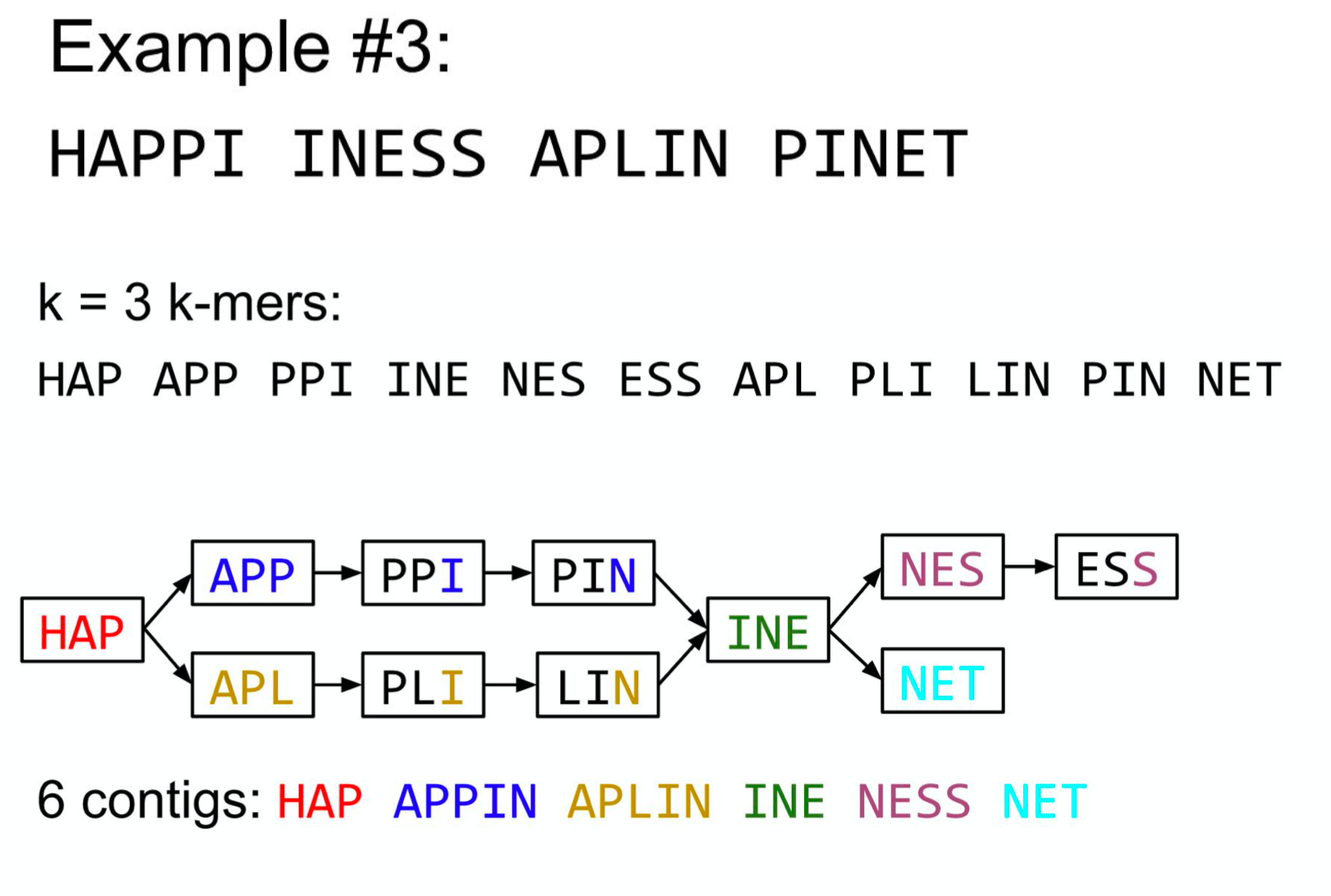
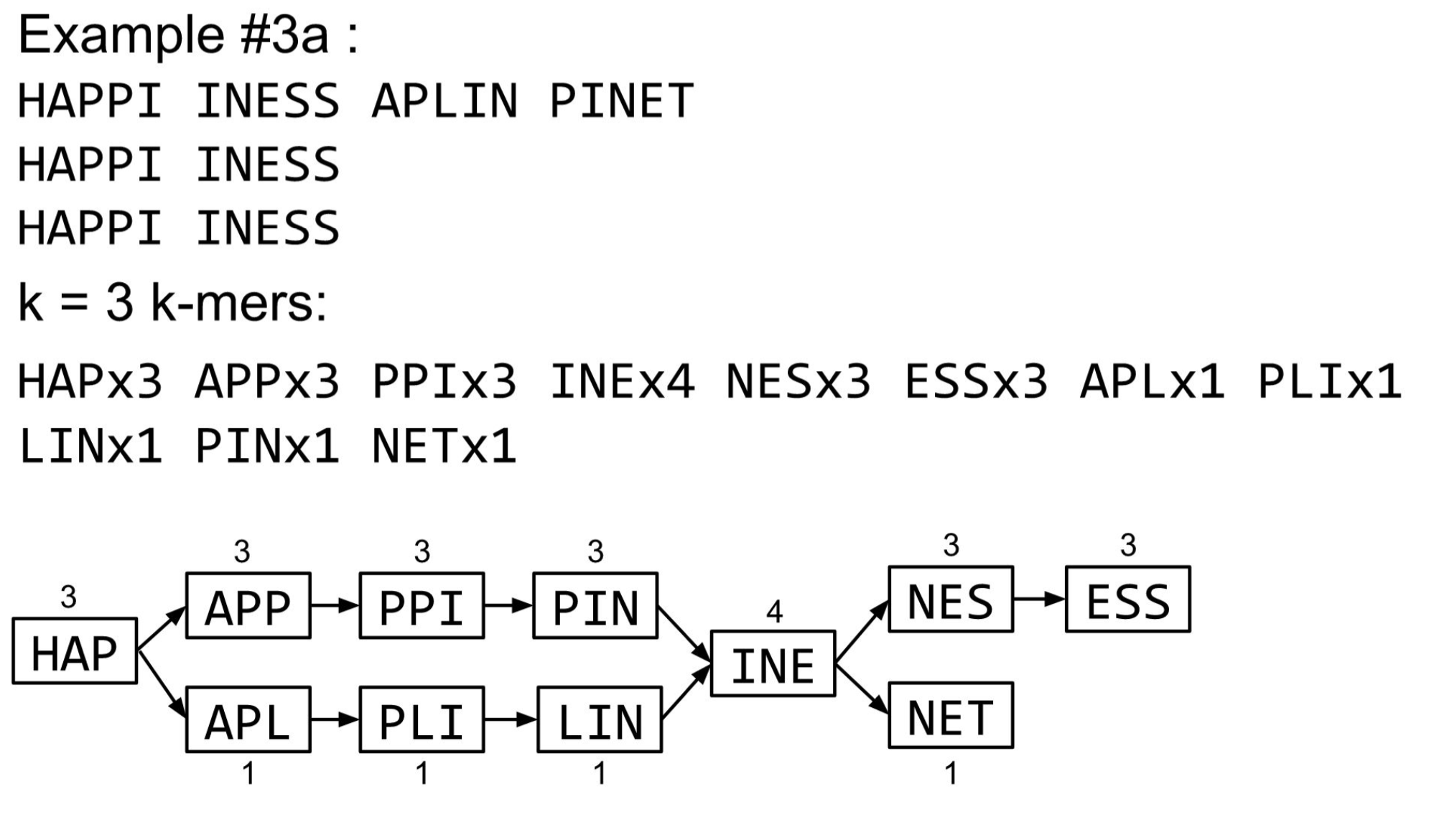
De Bruijn Graph
- A directed graph of sequences of symbols
- Nodes in the graph are k-mers
- Edges represent consecutive k-mers (k-1 symbols overlap)
Consider the 2 symbol alphabet (0 & 1) de Bruijn Graph for k=3
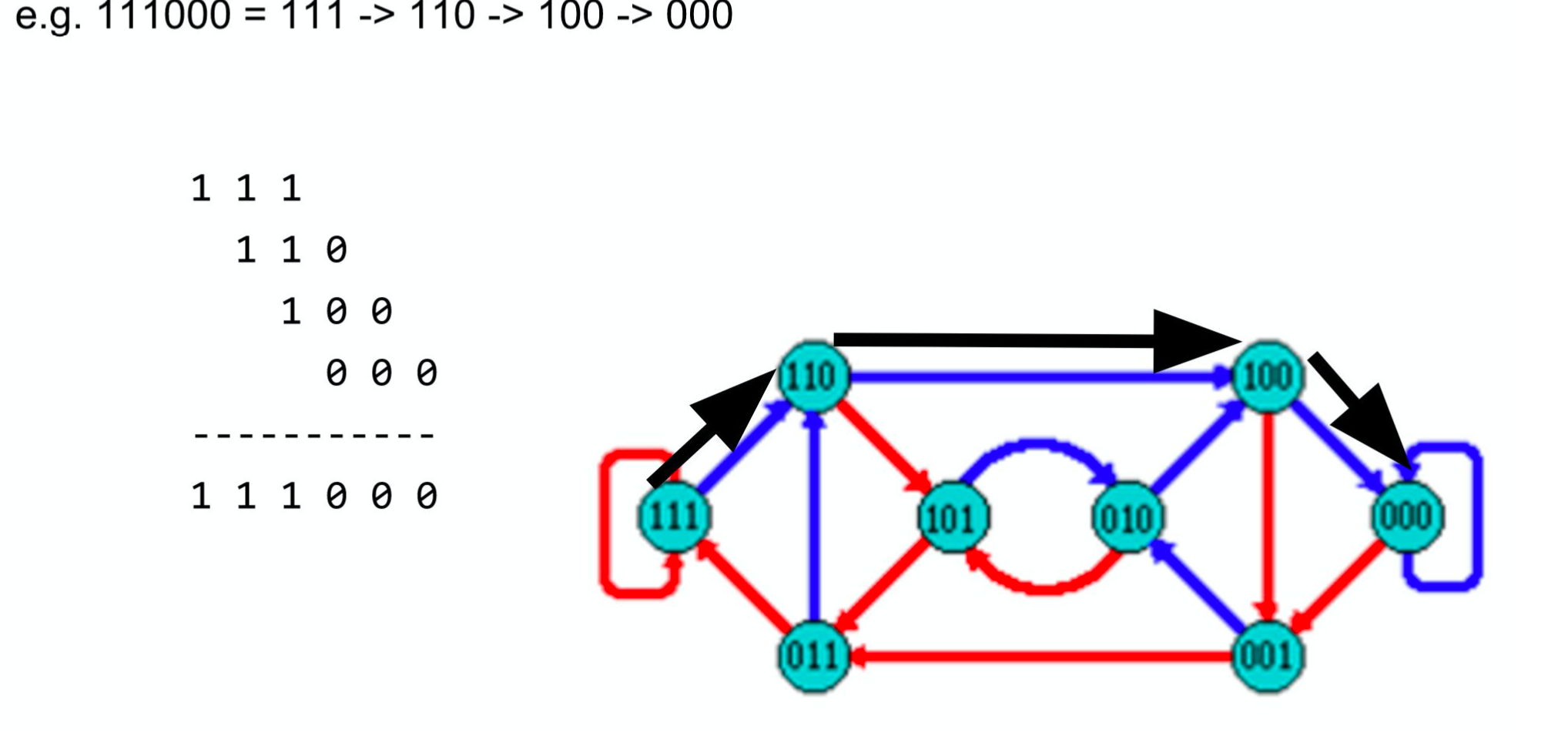
Producing sequences
- Sequences of symbols are produced by moving through the graph
What are K-mers ?
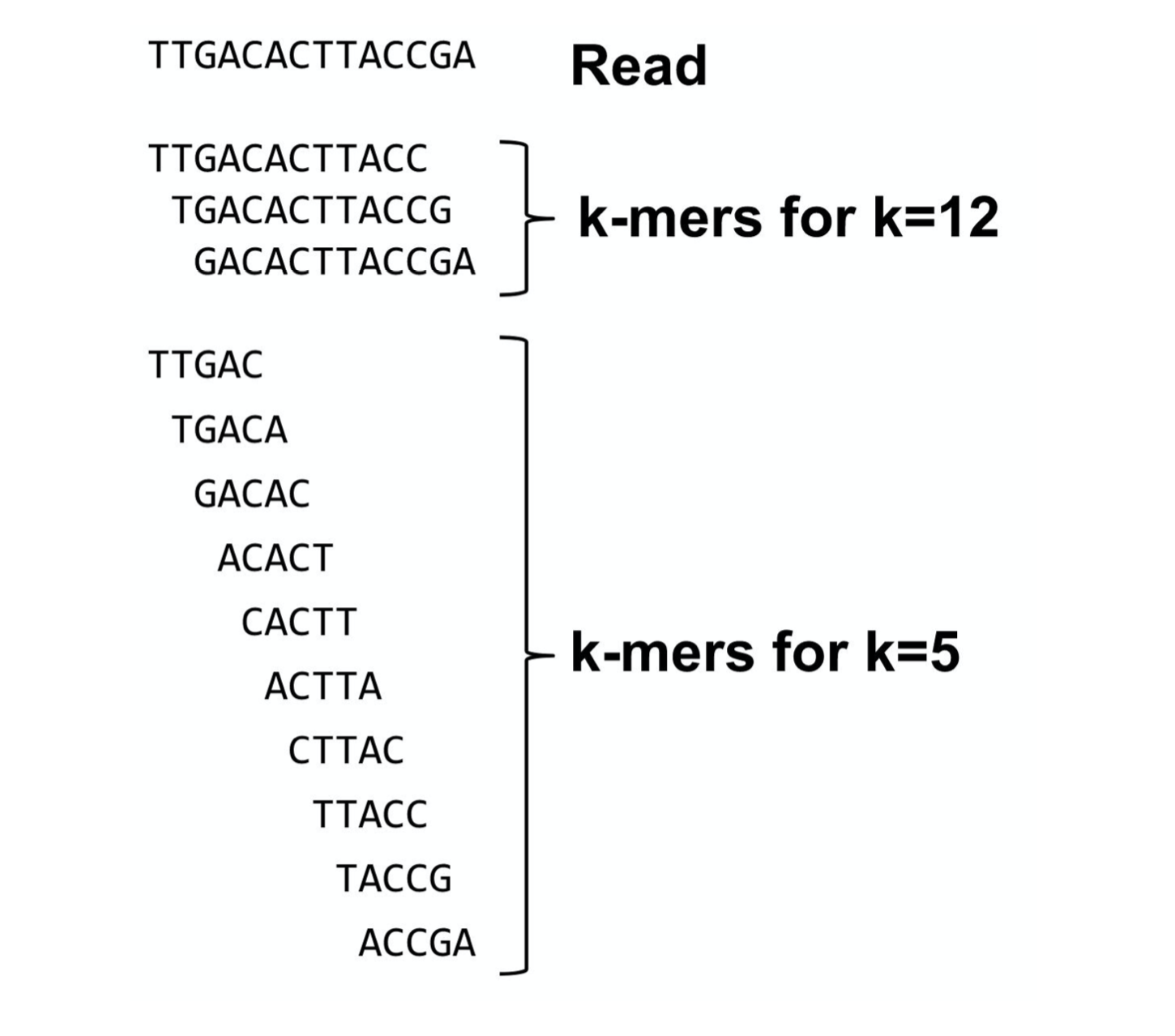
K-mers de Bruijn graph

K-mers de Bruijn graph

K-mers de Bruijn graph
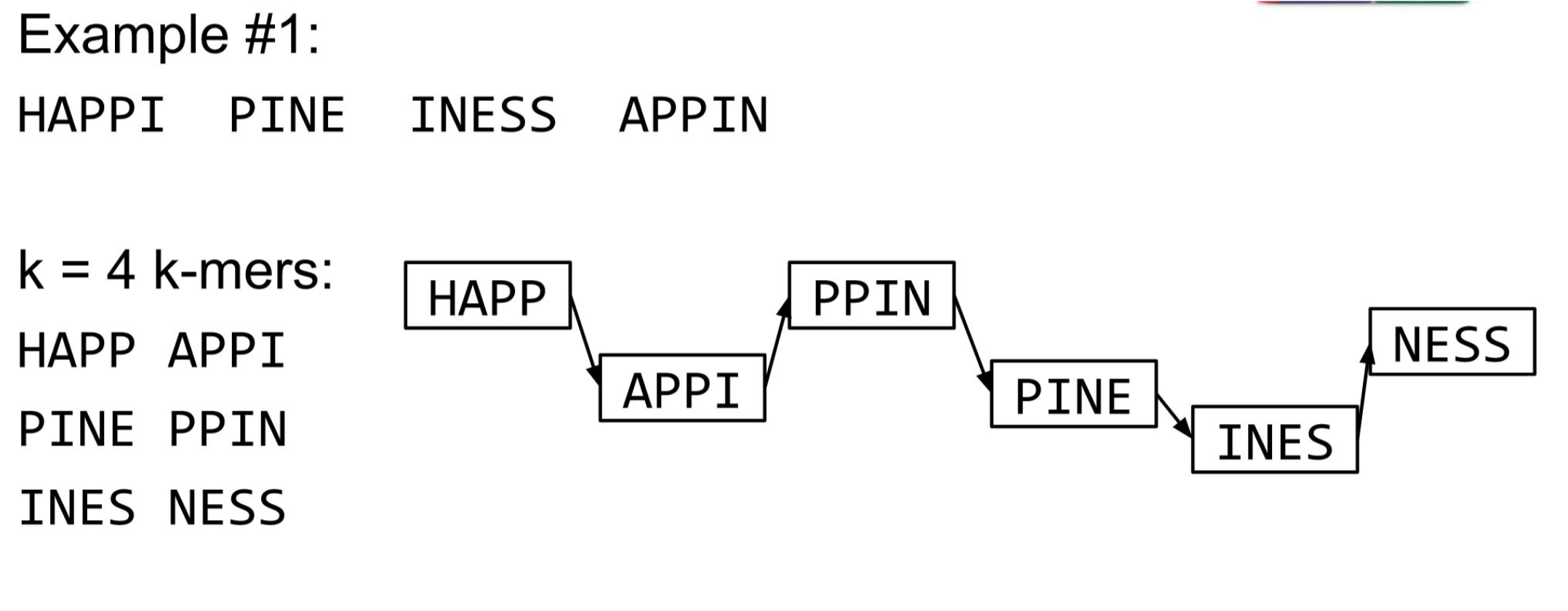
The problem of repeats

The problem of repeats
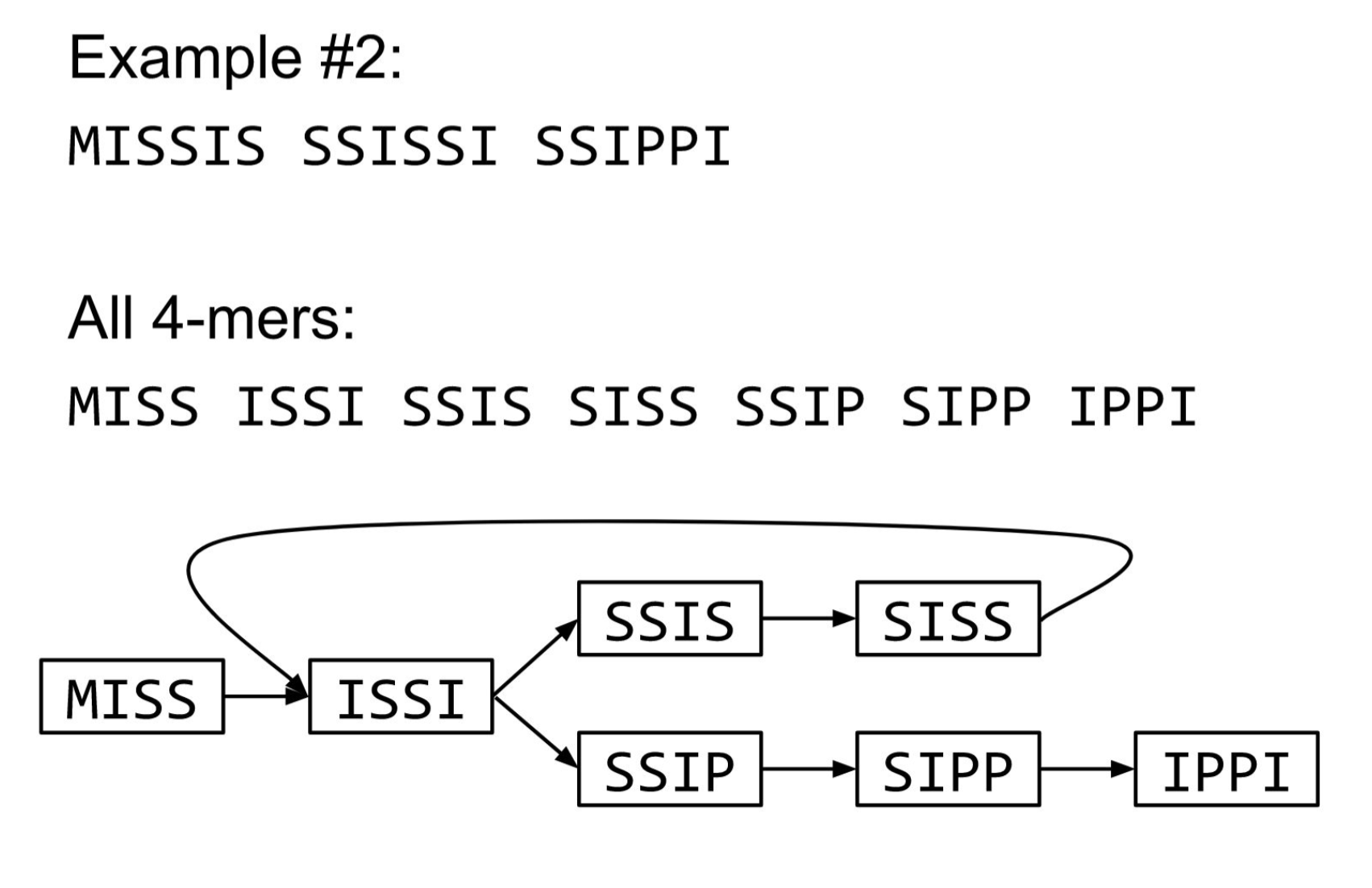
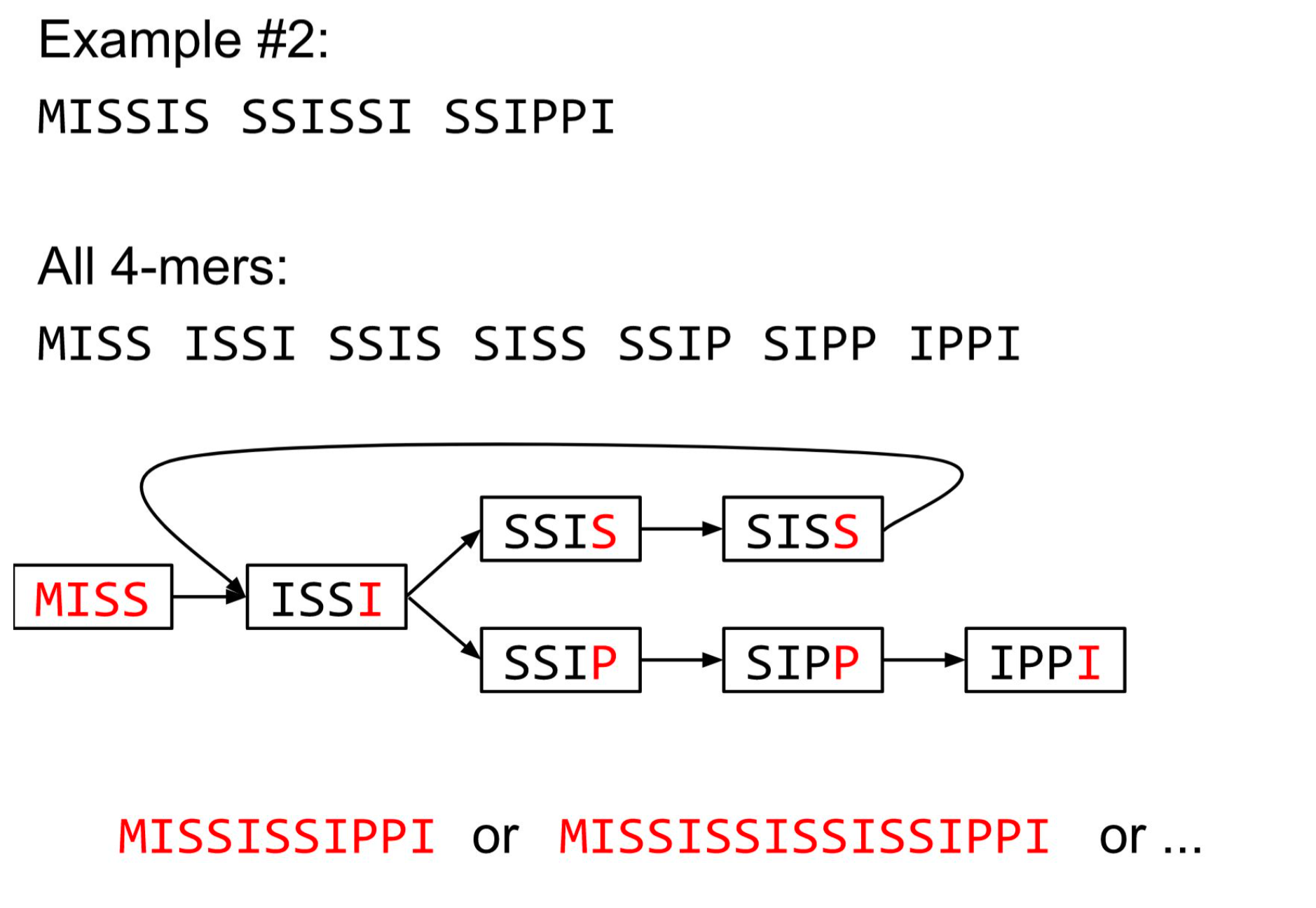
The problem of repeats
Different k

Different k
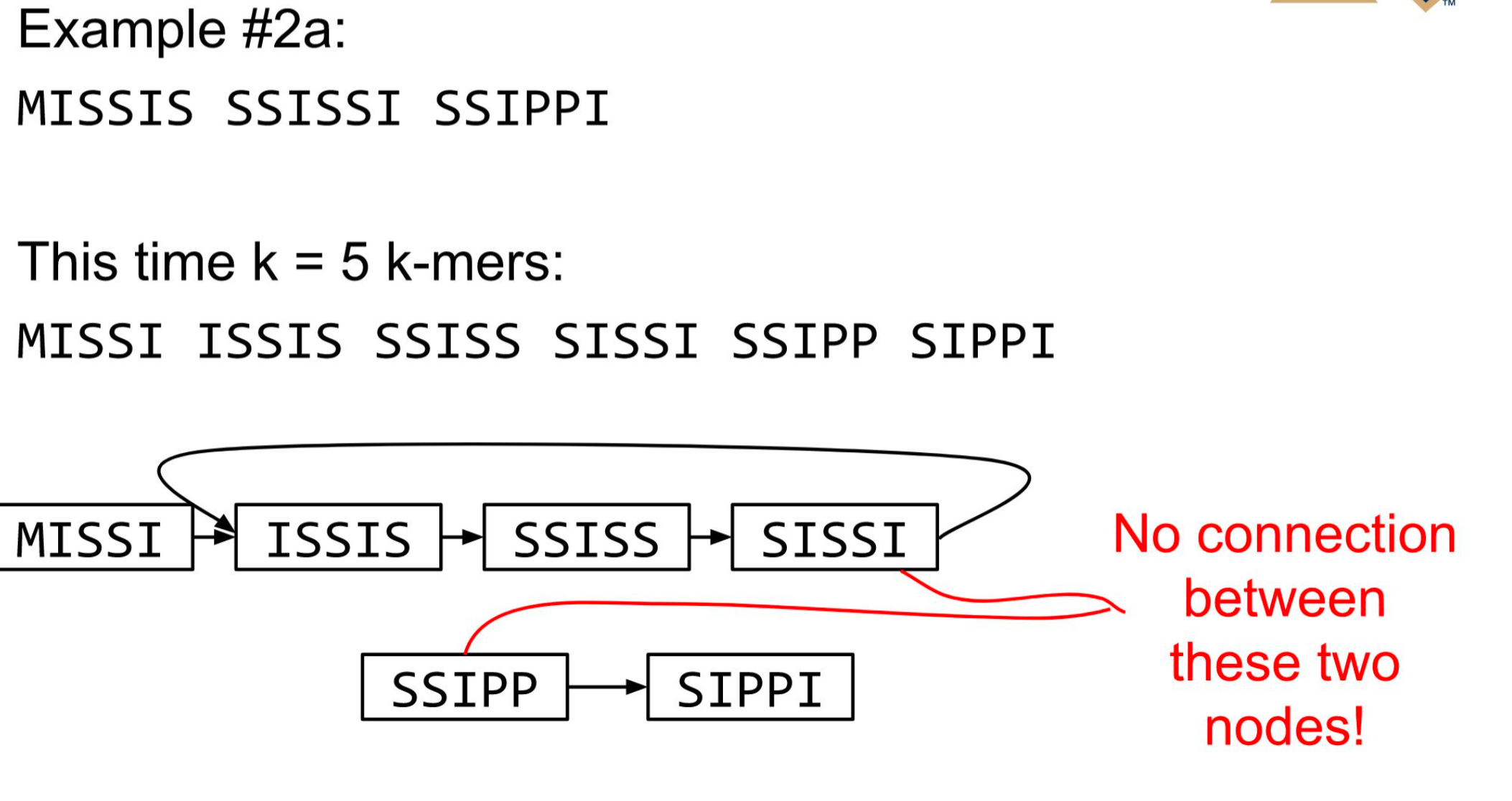
2 contigs : MISSISSIS & SSIPPI
Sequencing errors

Sequencing errors
Sequencing errors
Sequencing errors - coverage
Which path looks most valid ? Why ?
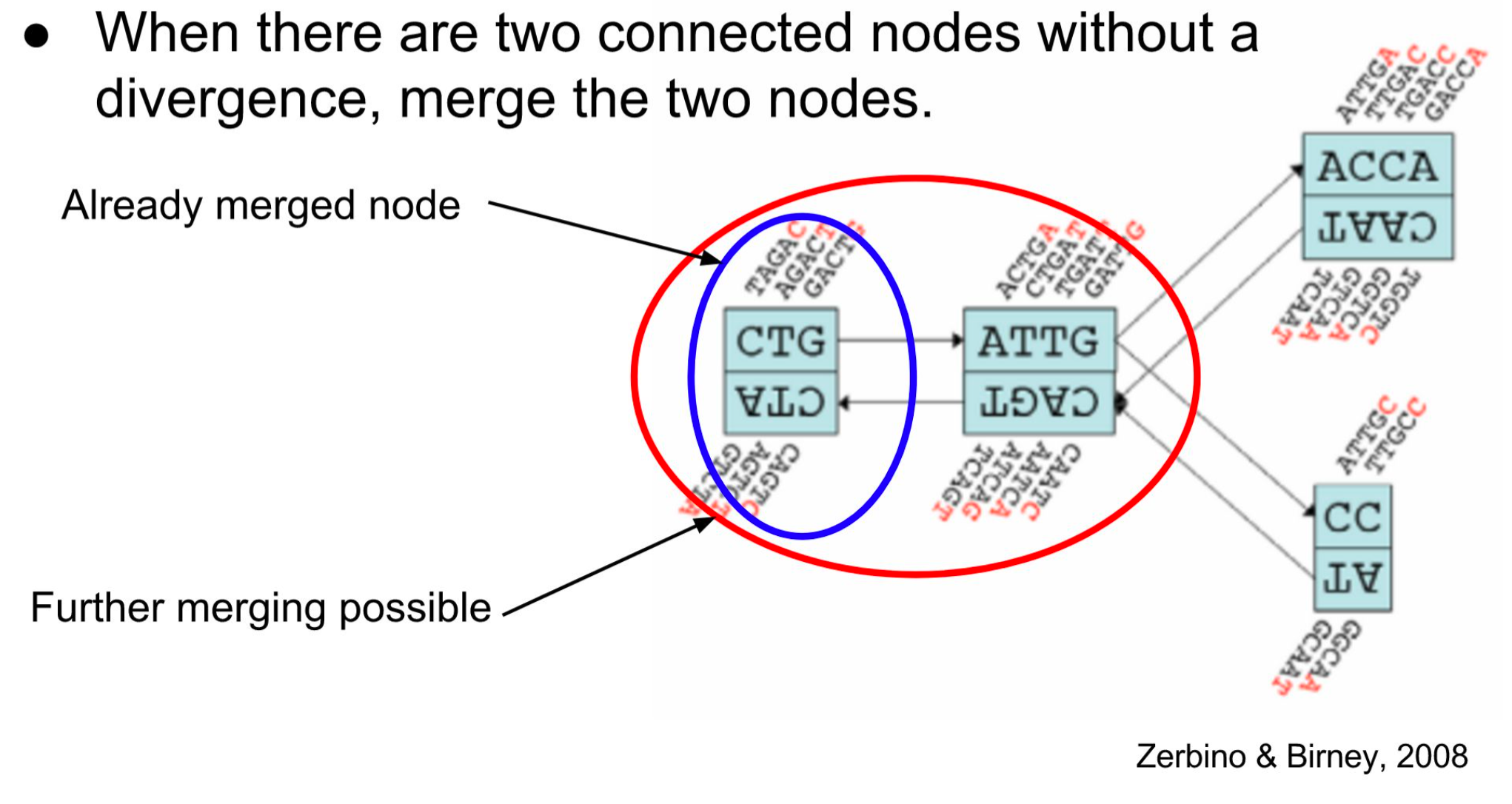
Graph simplification : Chain Merging
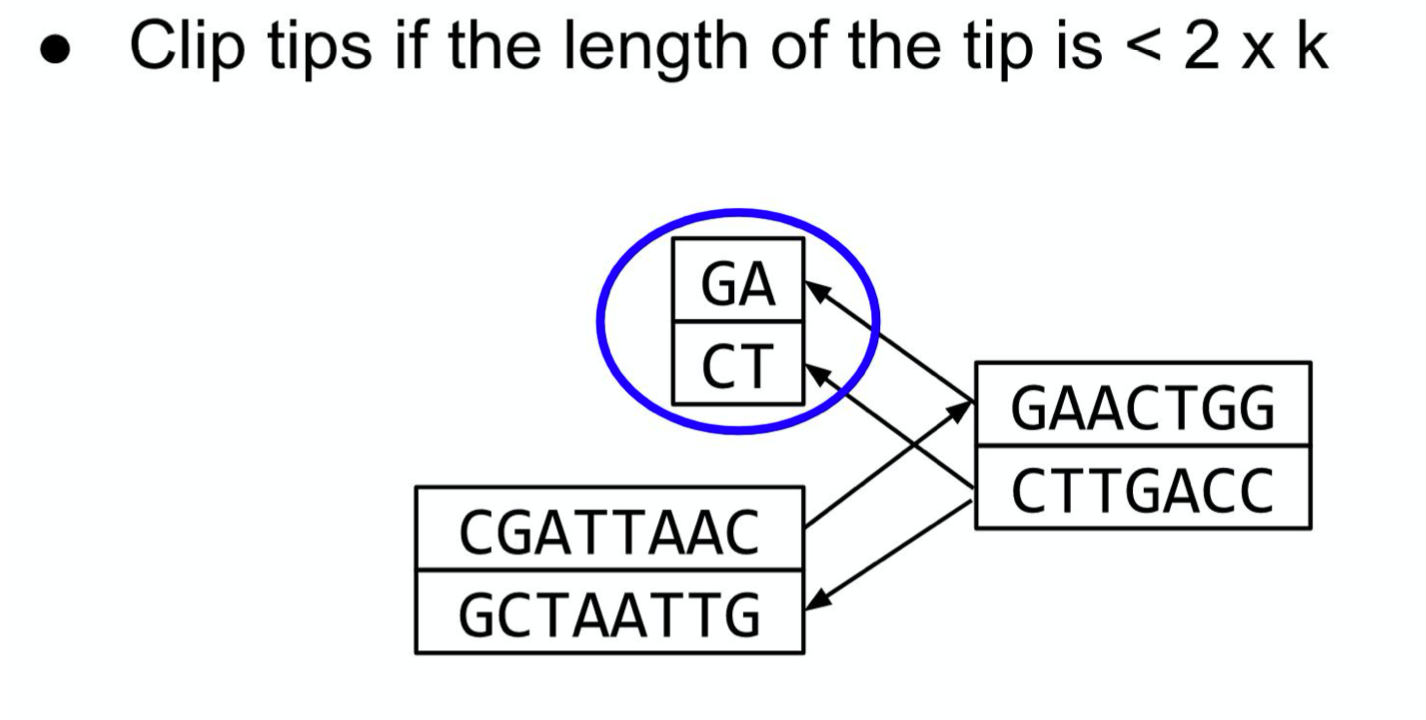
Graph simplification : Tip Clipping
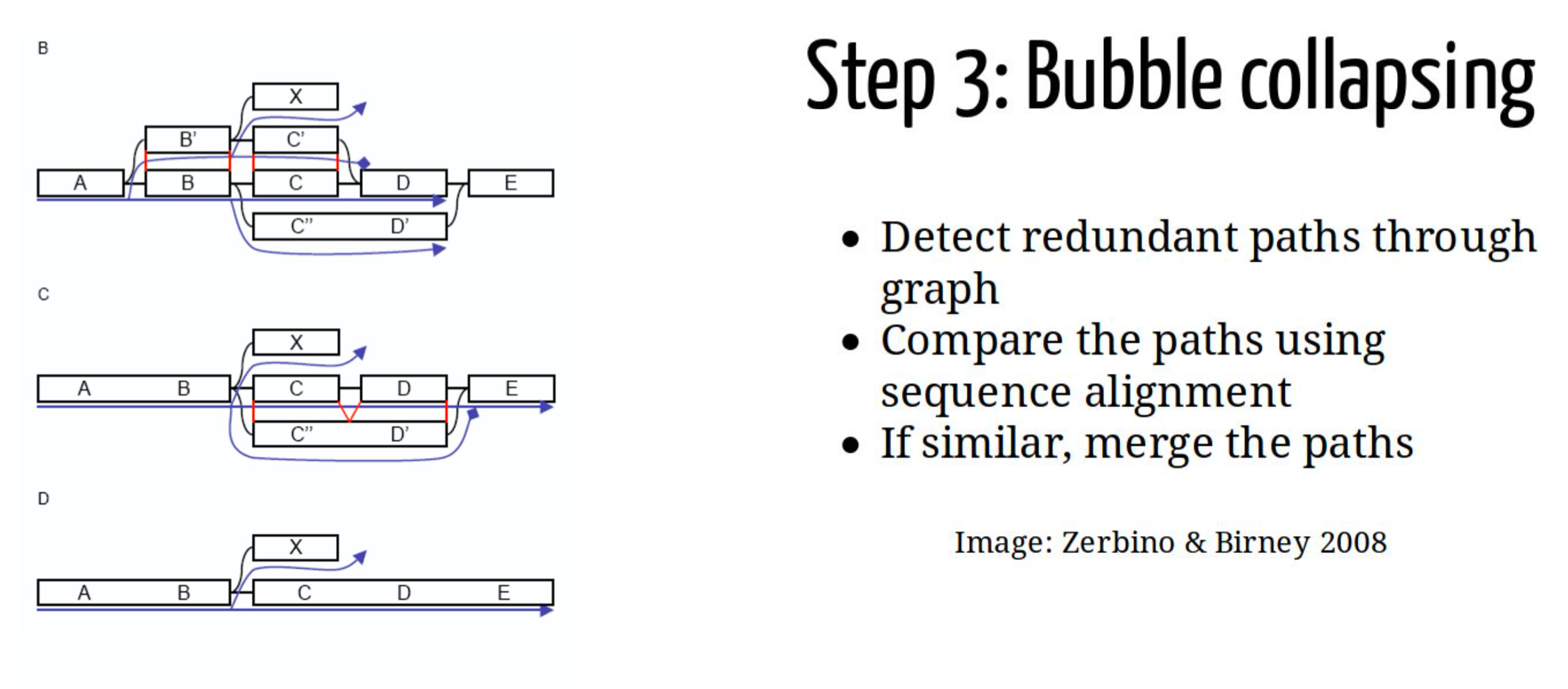
Graph simplification : Bubble Collapsing
Seed-and-extend for NGS data

Seed-and-extend for NGS data
Seed-and-extend mappers are a class of read mappers that break down each read sequence into seeds (i.e., smaller segments) to find locations in the reference genome that closely match the read.
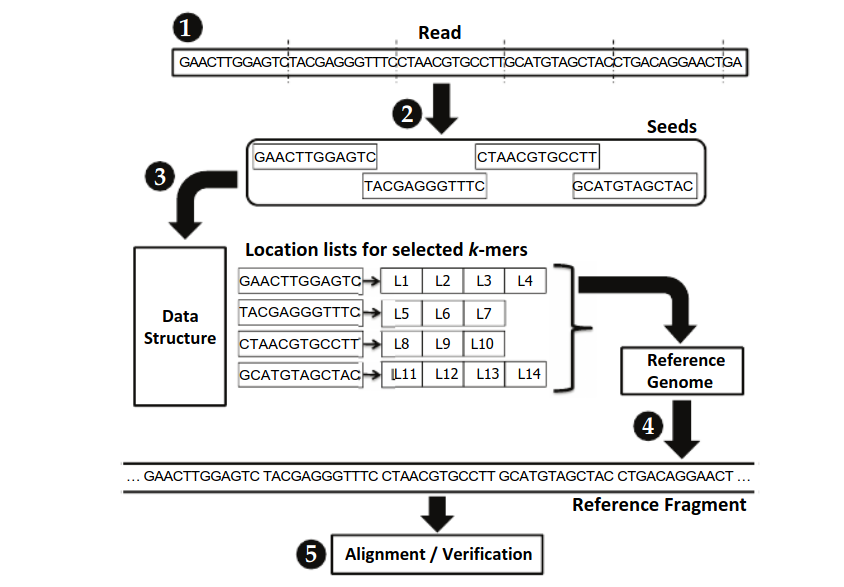
- The mapper obtains a read
- The mapper selects smaller DNA segments from the read to serve as seeds
- The mapper indexes a data structure with each seed to obtain a list of possible locations within the reference genome that could result in a match
- For each possible location in the list, the mapper obtains the corresponding DNA sequence from the reference genome
- The mapper aligns the read sequence to the reference sequence, using an expensive sequence alignment algorithm to determine the similarity between the read and the reference.
Mapping
- For further analysis it is necessary to map all the reads on the contigs.
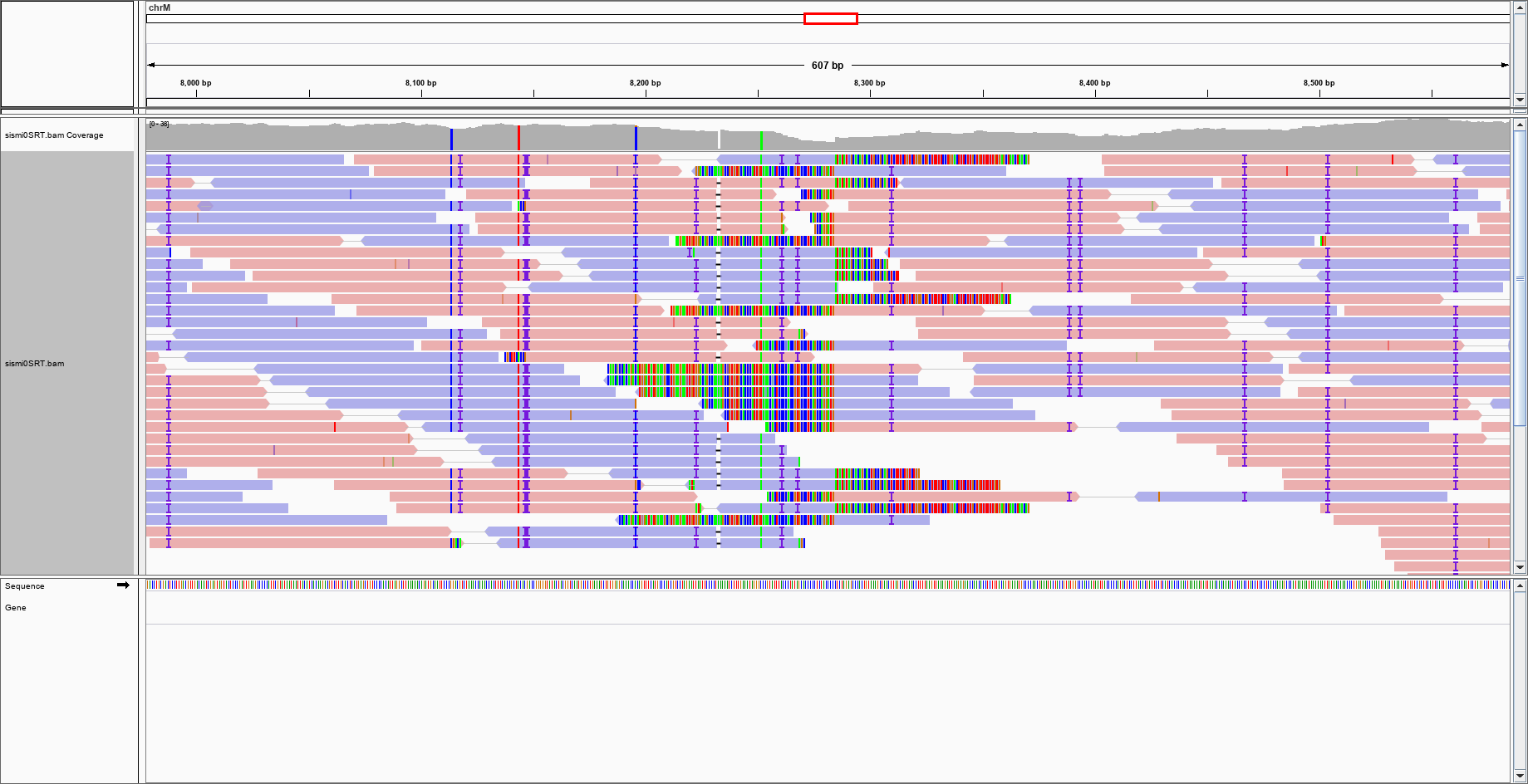
BAM/SAM

Visualization
- Some tools for visualization and browsing
- IGV (alignments and reference)
- Artemis (genome and annotations)