Easy16S est développée par Migale en Shiny et s’appuie largement sur le package {phyloseq}[2].
1.1 Disclaimer
Easy16S facilite l’exploration, la visualisation et l’analyse des données de métabarcoding. Cependant, les utilisateurs doivent veiller à ne pas sur‑interpréter les résultats. Une interprétation correcte des données métagénomiques nécessite une solide compréhension de l’écologie microbienne, de la biostatistique et du domaine d’étude spécifique. Bien que notre outil soit conçu pour être convivial, la complexité de l’analyse des données métagénomiques implique que les résultats peuvent être trompeurs s’ils ne sont pas évalués avec soin.
Nous avons intégré plusieurs paramètres par défaut et des garde‑fous pour guider les utilisateurs et réduire le risque d’utilisation abusive. Toutefois, si vous ne disposez pas de connaissances en métagénomique, il est fortement recommandé de collaborer avec des bioinformaticiens et des biostatisticiens afin de garantir des conclusions solides et fiables issues de vos travaux.
2 Datasets
Ce TP utilise les données du projet MetaPDOcheese[3]. Lors de ce projet, les communautés microbiennes de 44 AOP laitières françaises, divisées en 7 familles technologiques, ont été analysées. Les données brutes sont disponibles sur Recherche Data Gouv : 10.57745/UCJG6S.
Pour le TP, nous travaillerons avec 72 échantillons analysés avec FROGS [4] provenant de :
6 AOP : AOP1 à AOP6 (répartis en 3 Tech_family : Pâte persillée (PPS), Pâte pressée non cuite (PPNC) ou Pâte pressée cuite (PNC))
2 Season : Summer ou Winter
3 réplicats par AOP pour chaque saison
3 Import / Export des données
3.1 Importer les données dans Easy16S
Les données peuvent être importées dans l’application avec le bouton Select your data en haut à gauche de l’interface.
Demo : données d’exemple pour les TP et démonstrations
Input data : construire un objet phyloseq à partir de fichiers plats issus de FROGS (ou autres)
un fichier BIOM (format standard ou format FROGS )
un tableau tabulé de métadonnées (variables en colonnes, échantillons en lignes) ; le nom des échantillons (première colonne) doit être strictement identique à celui du fichier BIOM. Le délimiteur et le format des colonnes peuvent être spécifiés. Le délimiteur et le format des colonnes peuvent être spécifiés.
un arbre phylogénétique au format Newick
un fichier FASTA contenant les séquences représentatives
RData / RDS : importer un objet phyloseq déjà construit
Importez les données obtenues à l’issue du TP‑FROGS dans Easy16S en utilisant l’option Input data.
On peut définir le type des colonnes du tableau de métadonnées.
n : pour un nombre
f : pour un facteur (liste de modalités définies)
l : pour une valeur logique (TRUE/FALSE)
c : pour une chaine de caractères (privilégier les facteurs lorsque c’est possible)
D ou t ou T : pour une date, un temps, ou un datetime (date + temps) en ISO8601
Combien d’échantillons sont présents dans l’objet phyloseq ?
NoteSolution
72 échantillons
Combien de taxons (= ASVs) sont présents dans l’objet phyloseq ?
NoteSolution
398 taxons
Quelle est la profondeur de séquençage (nombre de reads) la plus faible ?
NoteSolution
La profondeur de séquençage la plus faible est de 6 989 reads.
3.3 Transformation et pré‑traitement des données
Avant de lancer les analyses statistiques, il est important de pré‑traiter les données, par exemple sélectionner les échantillons d’intérêt, modifier les comptes (raréfaction, normalisation, transformation, etc.). Cela se fait dans l’onglet Preprocess data. Les transformations s’appliquent de façon itérative à partir des données brutes.
Sélectionnez uniquement les échantillons des AOP AOP2 ou AOP3 puis raréfiez le résultat.
Combien d’échantillons et de taxons restent‑ils après le pré‑traitement ?
Après le pré‑traitement : 24 échantillons et 347 taxons.
L’historique des transformations appliquées est affiché dans .
Revenez aux données brutes avec le toggle.
Pour de nombreuses analyses, il est essentiel de travailler avec des données raréfiées afin d’éliminer l’effet de la profondeur de séquençage.
NoteSolution
physeq_rare <- physeq |>rarefy_even_depth(rngseed =314) # seed for Easy16S reproducibility
3.4 Export des données
Pour faciliter les analyses ultérieures, il est possible d’exporter les données courantes (brutes ou pré‑traitées selon le toggle) dans différents formats (.biom et .rds) via les boutons .
Exportez les données pré‑traitées au format .rds puis réimportez‑les dans une nouvelle session d’Easy16S.
4 Tables & Métadonnées
Toutes les tables sont triables, filtrables et exportables au format .csv.
L’onglet OTU/ASV Table présente la matrice d’abondance des ASVs par échantillon.
L’onglet Taxonomy Table donne l’affiliation taxonomique de chaque ASV.
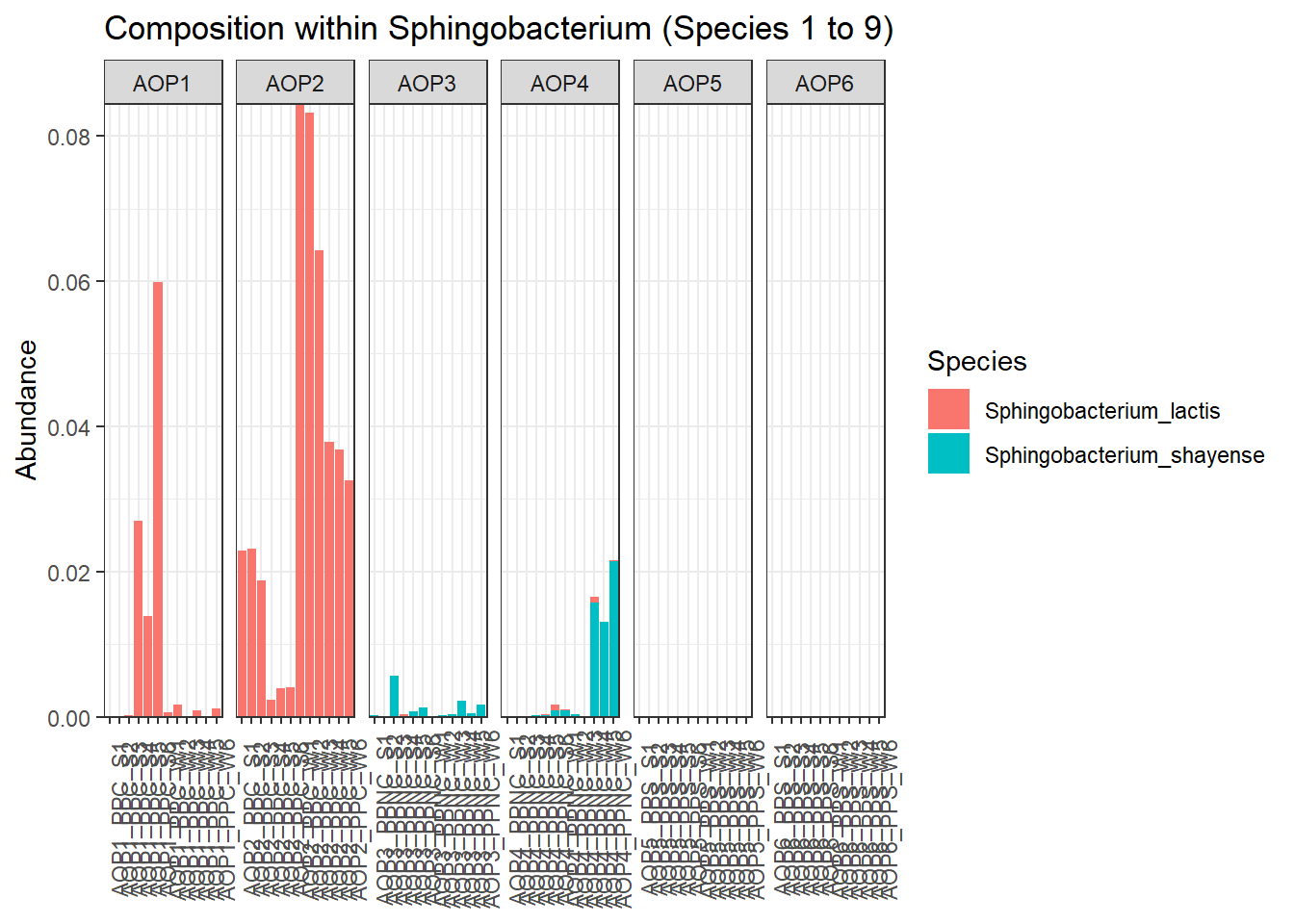
Quelle est l’affiliation taxonomique de l’ASV ID_278 ?
NoteSolution
ID_278 : Sphingobacterium lactis.
L’onglet Agglomerate ASV Table permet de visualiser la matrice d’abondance agrégée à un niveau taxonomique donné (ex. Genus).
L’onglet Sample Data Table contient les métadonnées associées à chaque échantillon.
Metadata permet de visualiser les métadonnées avec le package {esquisse} ; Ceci est utile pour explorer et visualiser les variables des échantillons (mais pas les données de métabarcoding).
5 Barplots
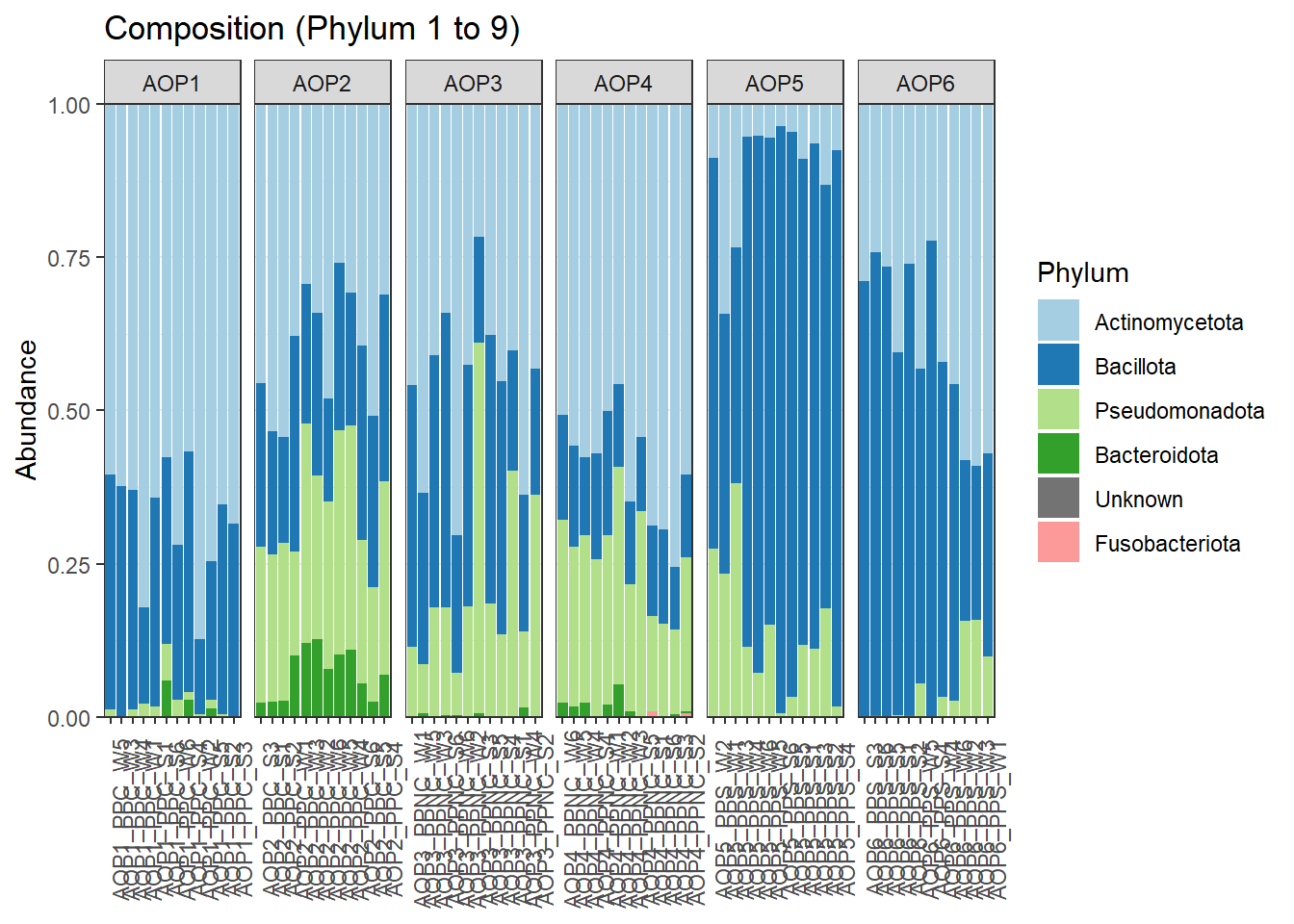
Le barplot d’abondance relative est la visualisation la plus courante. Dans Easy16S, vous pouvez afficher les abondances relatives de chaque échantillon à différents rangs taxonomiques via l’onglet Barplot. Il est possible de choisir un taxon d’intérêt (Selected filter taxa), définir le rang taxonomique utilisé pour la coloration (Taxonomic rank used for coloring), regrouper les échantillons selon une variable (Subplot), et ajuster l’ordre et le label des échantillons (Sample order / Sample label).
Visualisez l’ensemble des données au niveau Phylum en regroupant les échantillons par AOP et en les ordonnant selon le pH.
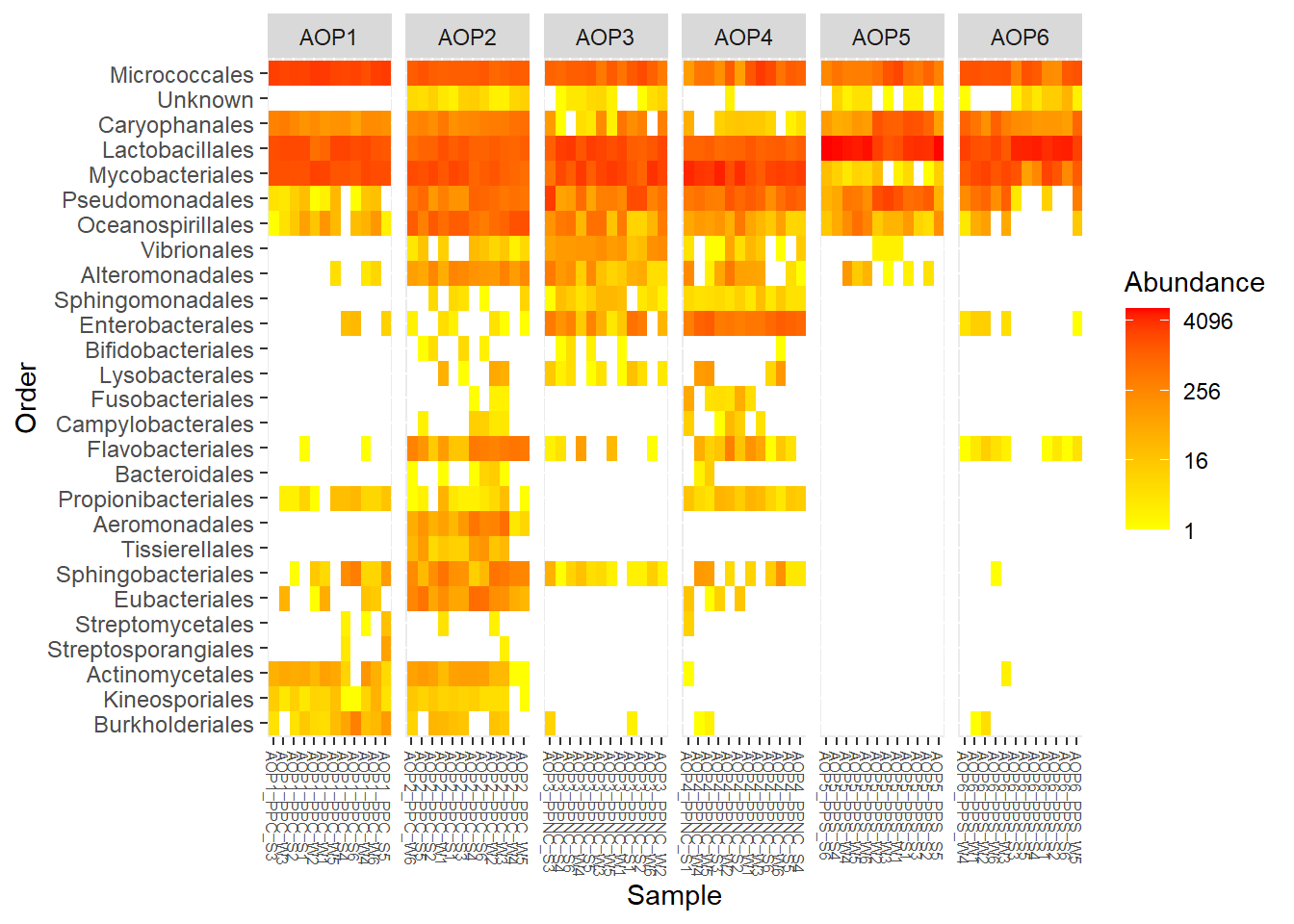
Avec l’onglet Heatmap, on peut afficher une carte de chaleur, permettant de visualiser l’abondance de chaque taxon dans chaque échantillon. Ceci permet d’observer les motifs structurants les communautés d’échatillons.
Les ASVs peuvent être regroupés à un rang taxonomique donnée. Les samples peuvent être regroupés, ordonnés et labelés en fonction des variables de métadonnées.
Il s’agit des données de comptage. Il faut s’assurer que les données sont normalisées ou raréfiées.
Affichez la structuration des communautés en agrégant au rang Order et en comparant les différents AOP.
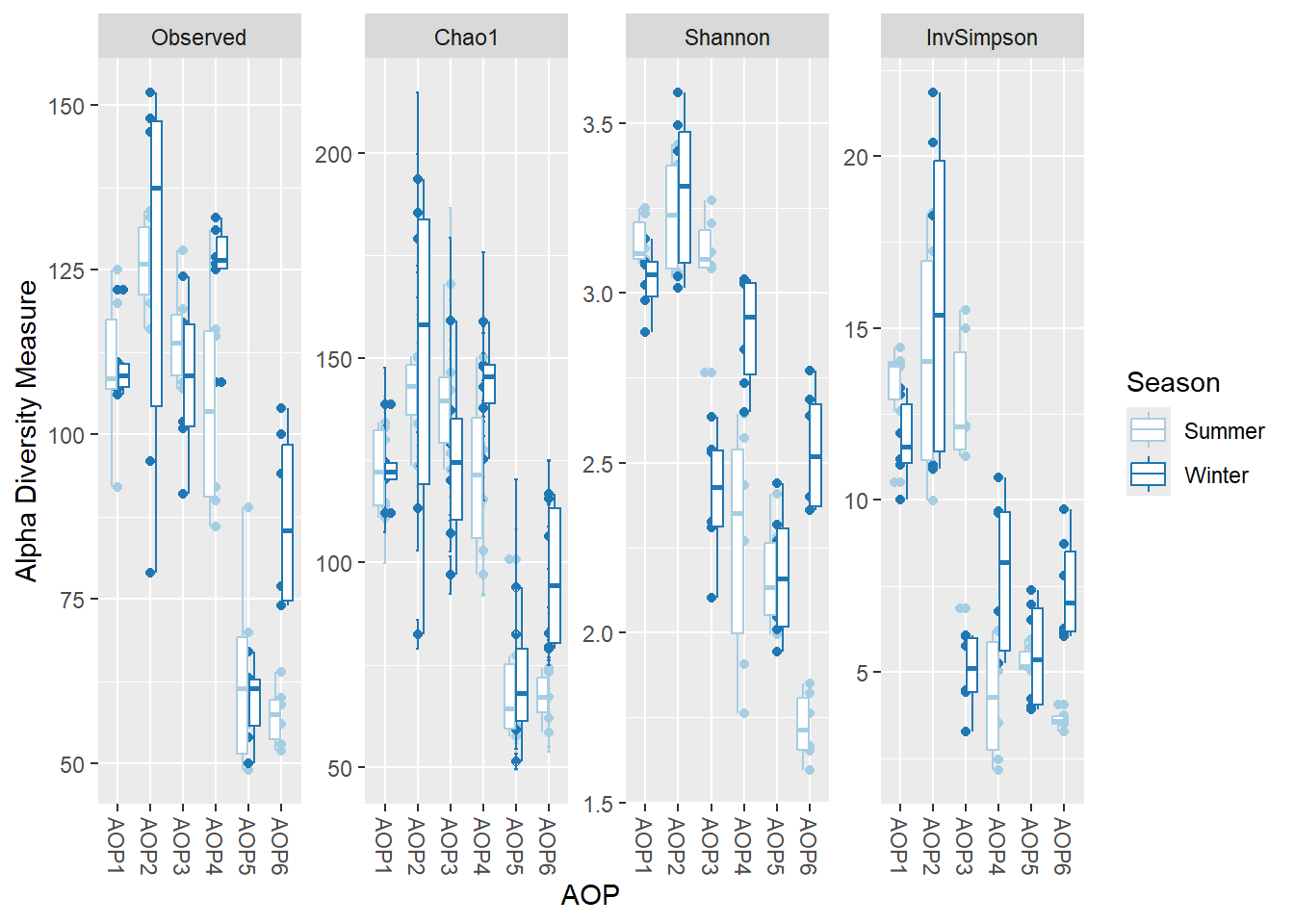
Cette section réalise une analyse de variance (ANOVA) sur une métrique de diversité en fonction d’une ou plusieurs variables de métadonnées, afin d’évaluer l’impact d’une covariable sur la richesse α.
Existe‑t‑il une corrélation significative entre la variable AOP et la richesse Chao1 ?
Analysis of Variance Table
Response: Chao1
Df Sum Sq Mean Sq F value Pr(>F)
AOP 5 55526 11105.3 26.771 1.116e-14 ***
Residuals 66 27378 414.8
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
9 β‑diversité
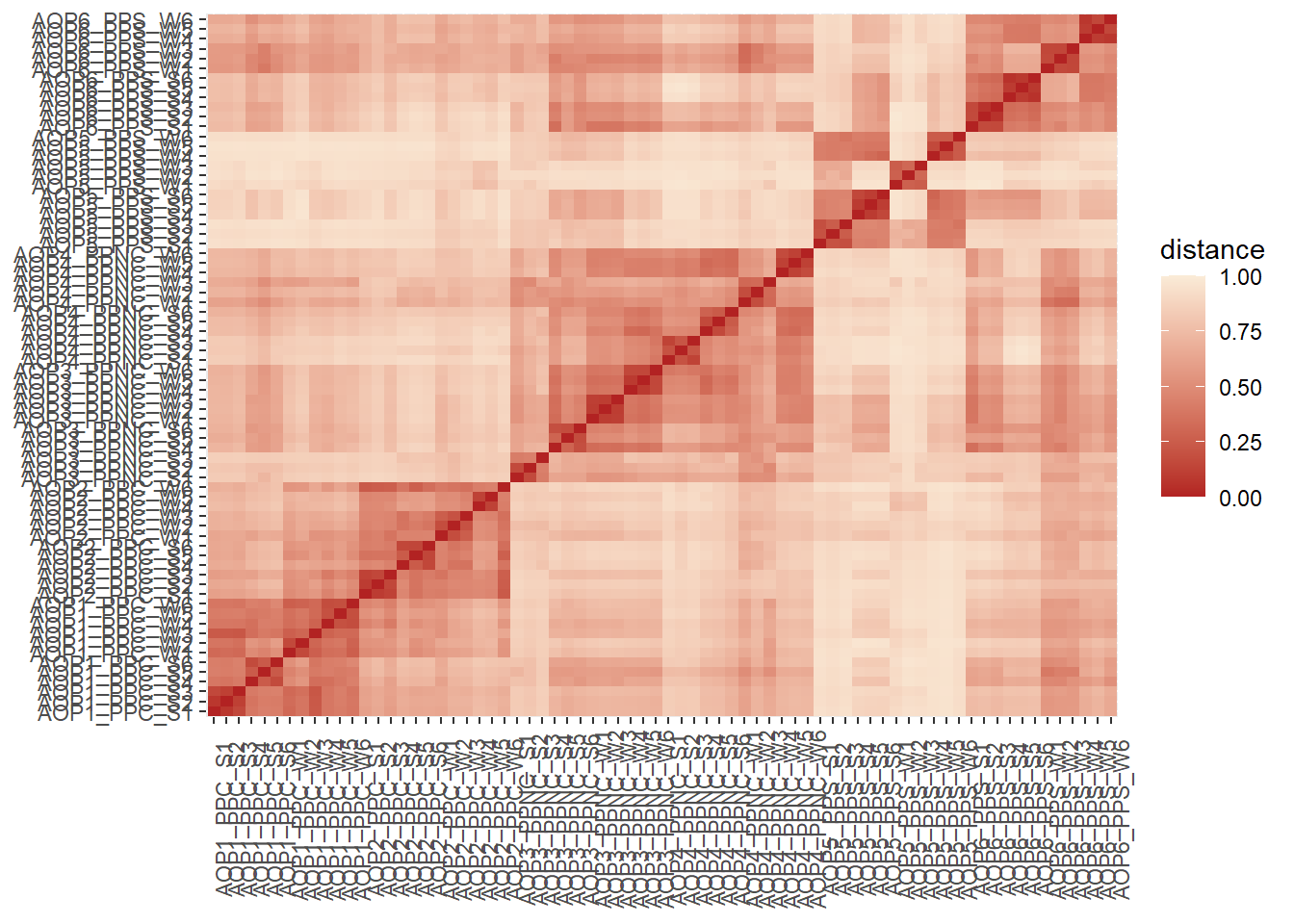
La β‑diversité mesure la dissimilarité entre les échantillons. On mesure donc la “distance” entre chaque paires d’échantillon. Plusieurs métriques existent s’appuyant sur des paramètres qualitatives ou quantitatives et phylogénétiques ou non. Le choix de la mesure est essentiel et influence fortement les résultats et leur interprétation.
Après avoir défini la distance à utiliser, on peut exploiter les résultats sous diverses forme :
Table des paires de distance
Heatmap de la matrice des distances
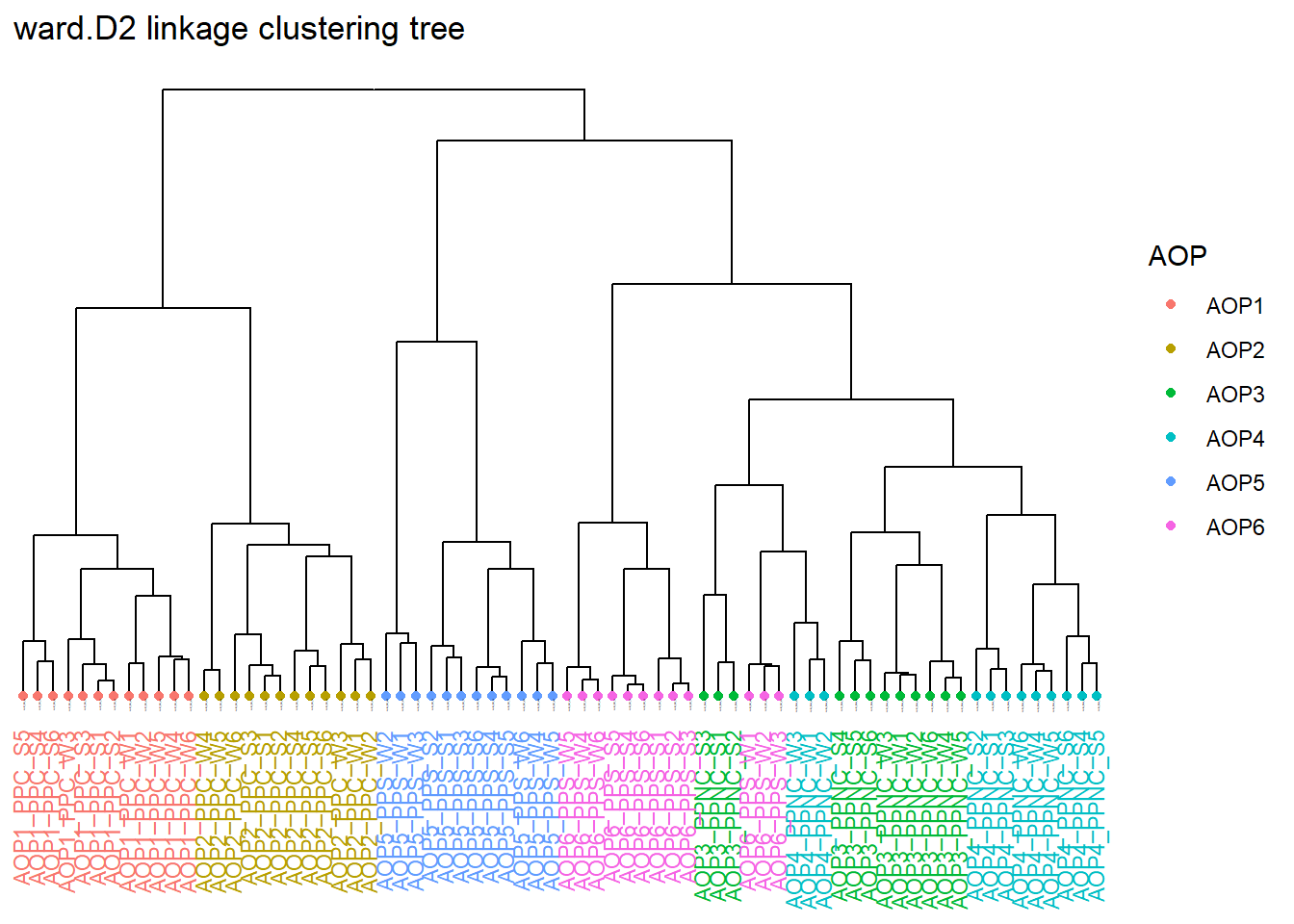
Dendogramme de clustering
MultiDimensional Scaling
Multivariate ANOVA
9.1 β ‑ Table
Pour chaque métrique, quels sont les deux échantillons les plus éloignés ?
Tracez le dendrogramme de clustering à partir des distances Bray‑Curtis et Jaccard, en coloriant les échantillons selon la métadonnée AOP.
NoteSolution
physeq_rare |>plot_clust(dist = dist_Bray, method ="ward.D2", color ="AOP")
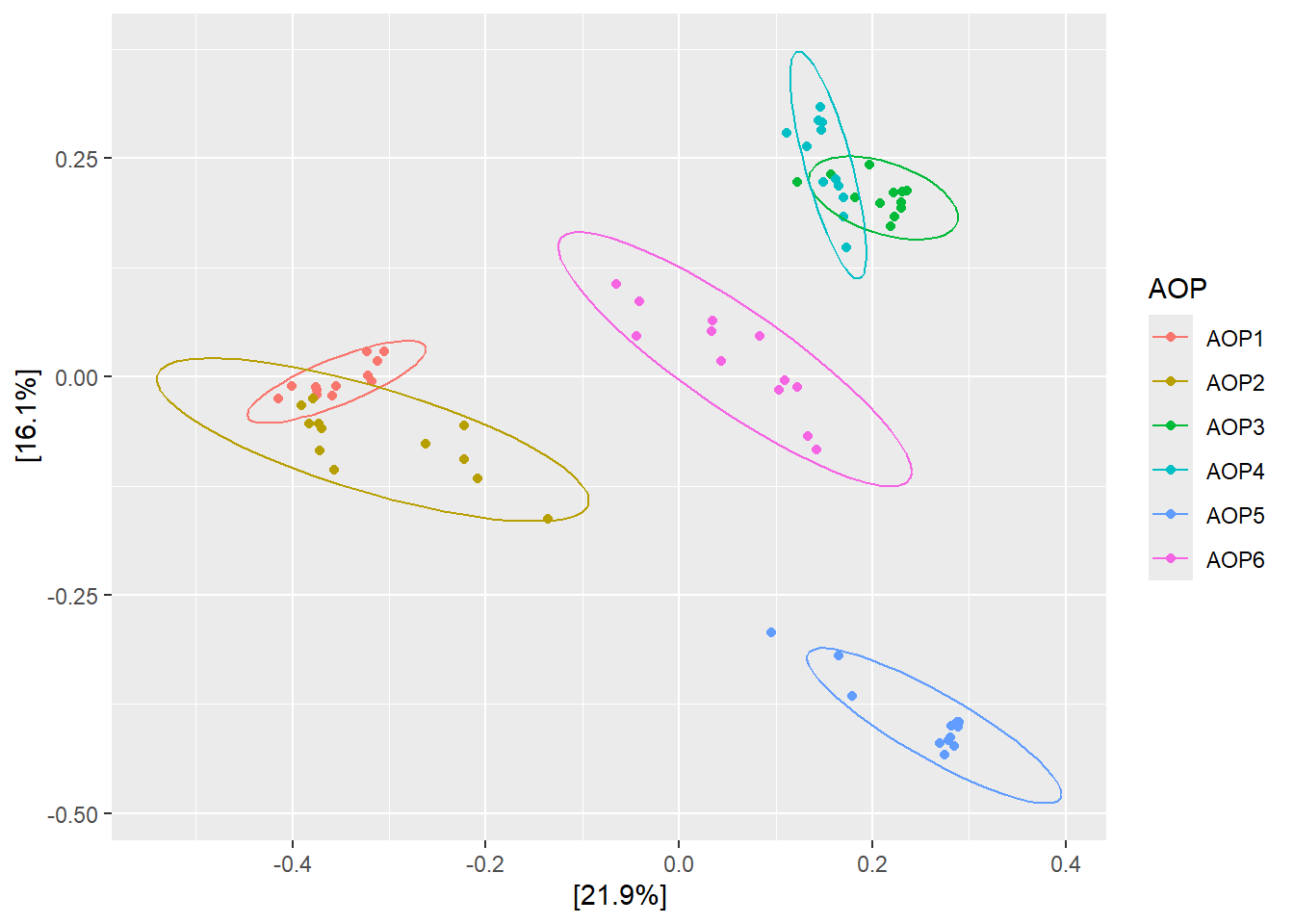
9.4 β ‑ MultiDimensional Scaling
De la même manière, il est possible de projeter le nuage de points des communautés sur un plan, en cherchant à préserver les distances entre les échantillons.
Projetez la matrice de distance de Jaccard (cc) avec la méthode d’ordination MDS, en coloriant les échantillons selon AOP.
Enfin, il est possible d’effectuer une analyse de variance de cette matrice de distance avec un test de permutation. On évalue ainsi l’impact d’une ou plusieurs covariables sur la structure de la communauté. Le test vegan::adonis2() compare la structure de nos données à 9999 structures générées par permutations aléatoires. La Permutational Multivariate ANOVA prend en charge les plans d’expérience complexes, mais elle ne teste que les effets localisé (telque, Est-ce que les communautés typiques sont similaires dans les groupes A et B ?) et suppose des dispersions égales (c’est-à-dire une variabilité biologique identique dans les deux groupes).
L’effet de l’AOP sur la matrice de distance Jaccard (cc) est‑il significatif ?
Permutation test for adonis under reduced model
Terms added sequentially (first to last)
Permutation: free
Number of permutations: 9999
vegan::adonis2(formula = dist_CC ~ AOP, data = metadata, permutations = 9999, by = "terms")
Df SumOfSqs R2 F Pr(>F)
AOP 5 11.0288 0.54946 16.098 1e-04 ***
Residual 66 9.0433 0.45054
Total 71 20.0721 1.00000
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
10 PCA
Bien que l’ordination MDS soit généralement privilégiée pour les données de microbiome, l’ACP peut être utilisée après une transformation appropriée des données.
Préférant les MDS, n’insisterons pas davantage sur les ACP dans ce TP.
11 Differential abundance
Après avoir constaté un effet significatif d’une covariable sur la structuration des communautés, on cherche souvant à identifier les ASVs sur‑ ou sous‑représentés selon une variable expérimentale donnée (catégorielle ou numérique). Le package {DESeq2}, développé à l’origine pour les RNA‑Seq, est couramment employé à cet effet.
Quels ASVs sont sur‑abondants ou sous‑abondants entre AOP3 et AOP5 ?
Ce jeu de données porte sur les communautés bactériennes présentes dans 8 matrices alimentaires différentes (EnvType), réparties en 4 produits carnés et 4 produits de la mer.
Quel est le microbiote caractéristique de chaque matrice ?
Comment les différentes matrices s’organisent‑elles ?
Cette étude explore les communautés bactériennes issues de milieux très variés (SampleType) afin d’analyser les structures écologiques à l’échelle mondiale.
Comment la profondeur de séquençage est‑elle distribuée ? Qu’est-il nécéssaire de mettre en place ?
Comparez les diversités α entre les environnements (SampleType). Quels environnements sont les plus ou les moins diversifiés ? Cette observation correspond‑elle à votre intuition ?
À partir des diversités β, que pouvez‑vous dire des différences entre les environnements ?
Les références
1. Midoux C, Rué O, Chapleur O, Bize A, Loux V, Mariadassou M. Easy16S: a user-friendly Shiny web-service for exploration and visualization of microbiome data. Journal of Open Source Software. 2024;9:6704. doi:10.21105/joss.06704.
2. McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS one. 2013;8:e61217.
3. Irlinger F, Mariadassou M, Dugat-Bony E, Rué O, Neuvéglise C, Renault P, et al. A comprehensive, large-scale analysis of « terroir » cheese and milk microbiota reveals profiles strongly shaped by both geographical and human factors. ISME Communications. 2024;4. doi:10.1093/ismeco/ycae095.
4. Bernard M, Rué O, Mariadassou M, Pascal G. FROGS: a powerful tool to analyse the diversity of fungi with special management of internal transcribed spacers. Briefings in Bioinformatics. 2021;22. doi:10.1093/bib/bbab318.
5. Chaillou S, Chaulot-Talmon A, Caekebeke H, Cardinal M, Christieans S, Denis C, et al. Origin and ecological selection of core and food-specific bacterial communities associated with meat and seafood spoilage. The ISME Journal. 2014;9:1105‑18. doi:10.1038/ismej.2014.202.
6. Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences. 2010;108:4516‑22. doi:10.1073/pnas.1000080107.
Réutilisation
CC BY‑SA
A work by Migale Bioinformatics Facility
Université Paris-Saclay, INRAE, MaIAGE, 78350, Jouy-en-Josas, France
Université Paris-Saclay, INRAE, BioinfOmics, MIGALE bioinformatics facility, 78350, Jouy-en-Josas, France