

cd MyDir # les fichiers FASTQ sont dans ce répertoire
tar zcvf Archive.tar.gz *.fastq.gzCe document vous présente la démarche et la réflexion pour analyser un jeu de données de métabarcoding avec l’outil FROGS [1]. Les analyses ont été effectuées sur l’instance Galaxy de l’IFB avec la version 5.1.0+galaxy0.
Avertissement
Les réponses aux questions de ce TP peuvent être visibles en appuyant sur le bouton Solution. À n’utiliser que si vous bloquez bien sûr !
1 Introduction à Galaxy
1.1 Instance Galaxy de l’IFB

Rendez-vous sur l’instance Galaxy de l’Institut Français de Bioinformatique : https://usegalaxy.fr. Puis allez sur cette page :
Cela vous permettra d’être associé à des ressources dédiées, allouées spécifiquement pour cette formation.
1.2 TP de découverte / rappels
Galaxy propose des tutoriels pour découvrir l’interface ou apprendre à utiliser certains outils. Ce tutoriel vous explique comment utiliser l’interface, importer des données et comment les organiser.
- Connectez-vous sur l’instance
NoteSolution
- Explorez et comprenez les différents panels de Galaxy
Nous allons suivre ces slides de présentation : https://galaxyproject.github.io/training-material/topics/introduction/tutorials/introduction/slides.html
- Créez votre premier historique nommé TP FROGS 16S 2026
NoteSolution
Note
Vous devez maintenant être capable de :
2 Contrôle qualité des données de séquençage
Nous allons analyser la qualité de 2 échantillons, soit 4 fichiers FASTQ. Bien sûr, pour vos données, vous devrez analyser tous vos échantillons.
- Sur votre ordinateur, téléchargez l’archive à l’adresse
http://genome.jouy.inra.fr/~orue/FROGS_2026/MPC_16S.tar.gz - Décompressez l’archive
- Uploadez 4 fichiers FASTQ (ceux que vous souhaitez) dans l’historique Galaxy TP FROGS 16S 2026
NoteSolution
- Lancez FastQC
[2] pour analyser la qualité des échantillons
Avertissement
Il est possible de choisir plusieurs datasets en même temps pour leur appliquer la même action ou de créer une collection !
NoteSolution
- Lancez MultiQC
[3] pour obtenir une rapport synthétique
NoteSolution
Explorez le rapport HTML généré
Quel est le principal atout de cet outil ?
NoteSolution
MultiQC
- Est-ce que tous les reads sont de la même longueur ? Qu’est-ce que cela signifie ?
NoteSolution
Oui, les reads sont tous de longueur 251 bp. Cela indique qu’aucun traitement n’a été effectué sur les données brutes. Si les reads ne sont pas tous de la même longueur pour un séquençage Illumina ou AVITI, il faut comprendre pourquoi et contacter la plateforme de séquençage.
- La qualité des bases vous semble-t-elle correcte ?
NoteSolution
Les données de séquençage Illumina ont généralement des profils équivalents, avec une qualité de base qui se dégrade vers la fin du read.
3 Préparer une archive pour FROGS
FROGS a besoin que tous les fichiers d’entrée lui soient fournis, mais contrairement aux collections, l’outil ne doît pas être lancé sur chaque fichier. Sans archive, il faut donc entrer tous les échantillons les uns après les autres. C’est fastidieux et peut causer des erreurs.
Une archive est une sorte de fichier qui contient plein de fichiers.
Cette archive doit avoir les propriétés suivantes :
- Elle ne doit contenir que les fichiers FASTQ, pas de dossiers ni d’autres fichiers
- Les fichiers FASTQ peuvent être compressés
- Les fichiers FASTQ doivent être suffixés, si les données sont pairées, par *_R1.fastq.gz* et *_R2.fastq.gz*
- Le nom de l’échantillon est construit à partir des chaines de caractères précédant ces suffixes.
En ligne de commande :
Ce document explique comment créer une archive sous Windows.
4 Analyse du jeu de données 16S
Il s’agit de résultats issus du travail de Irlinger et al., 2024 s’intéressant aux communautés bactériennes et fongiques de fromages AOP français.


Important
Le jeu de données a été récupéré sur les banques publiques puis a été sous-échantillonné et réduit pour permettre un temps d’analyse raisonnable pour la formation. La procédure détaillée est disponible ici
Nous avons gardé 72 échantillons 16S et 72 échantillons ITS avec les caractéristiques suivantes :

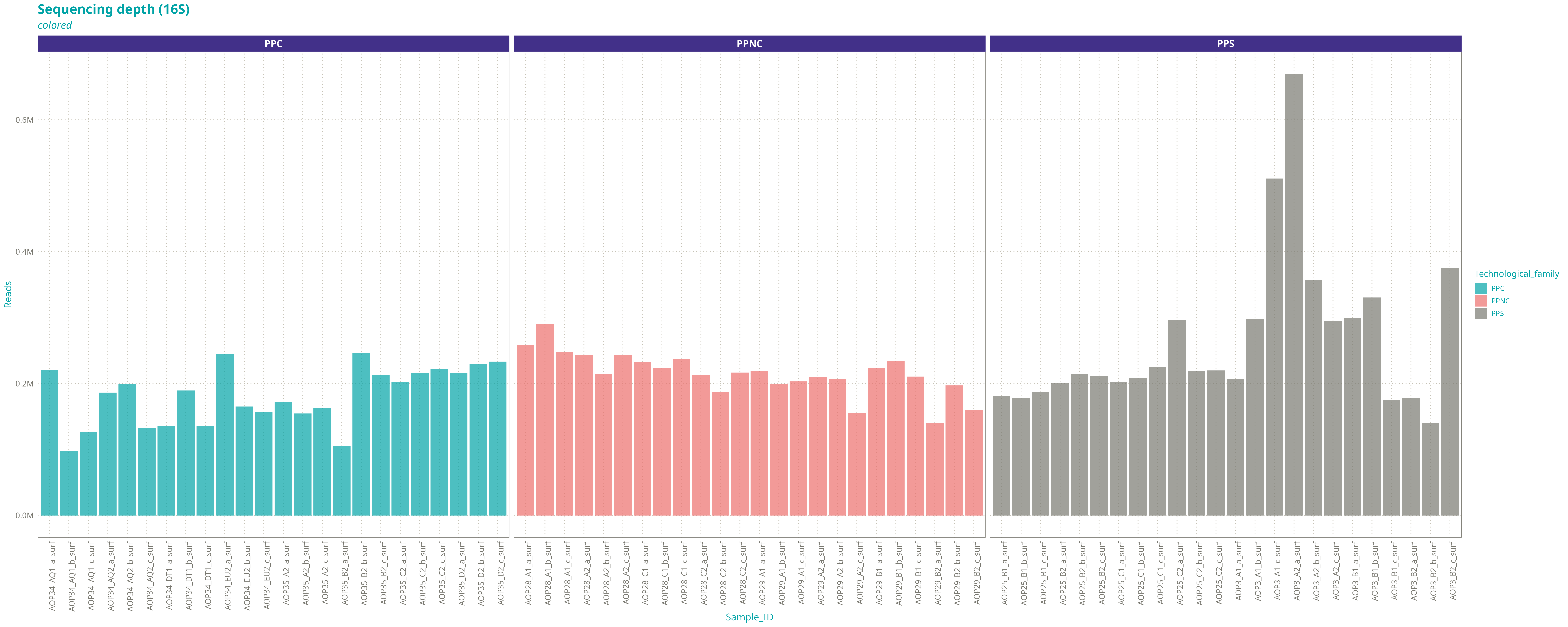
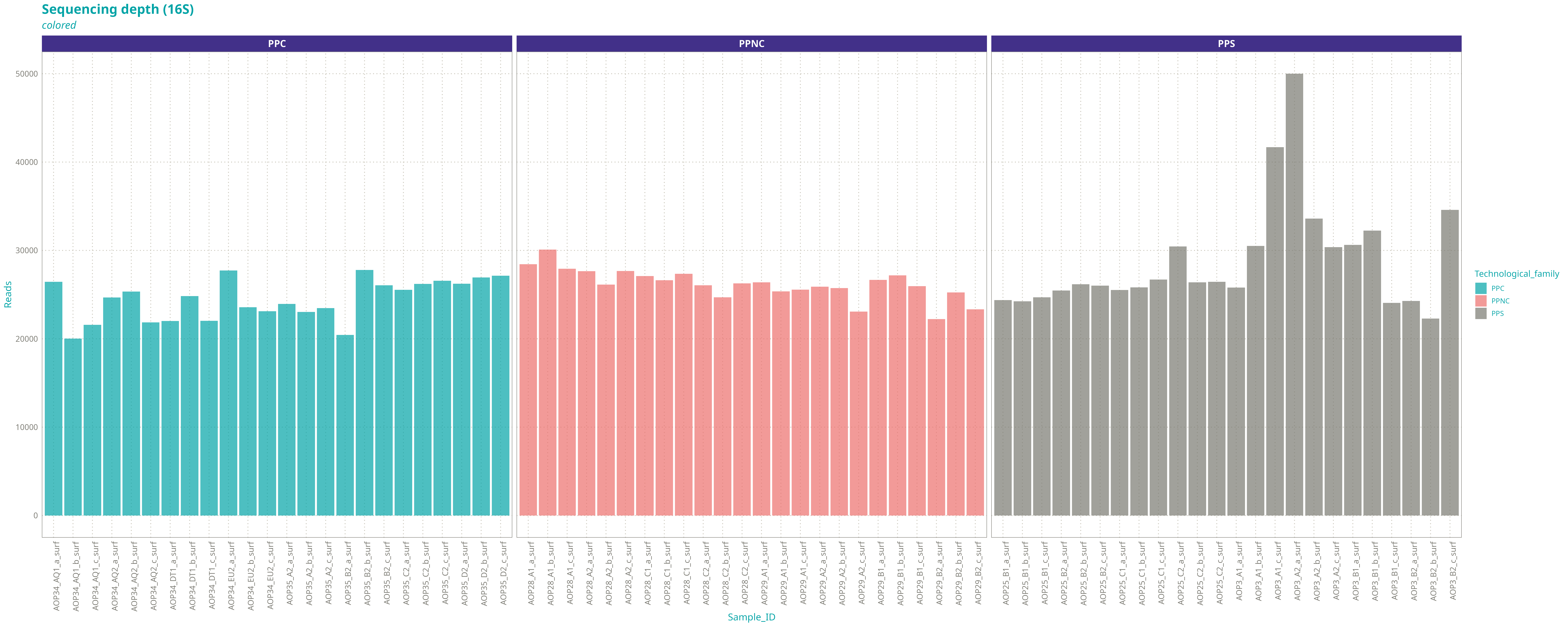
Nombre de reads du jeu réel / du jeu réduit
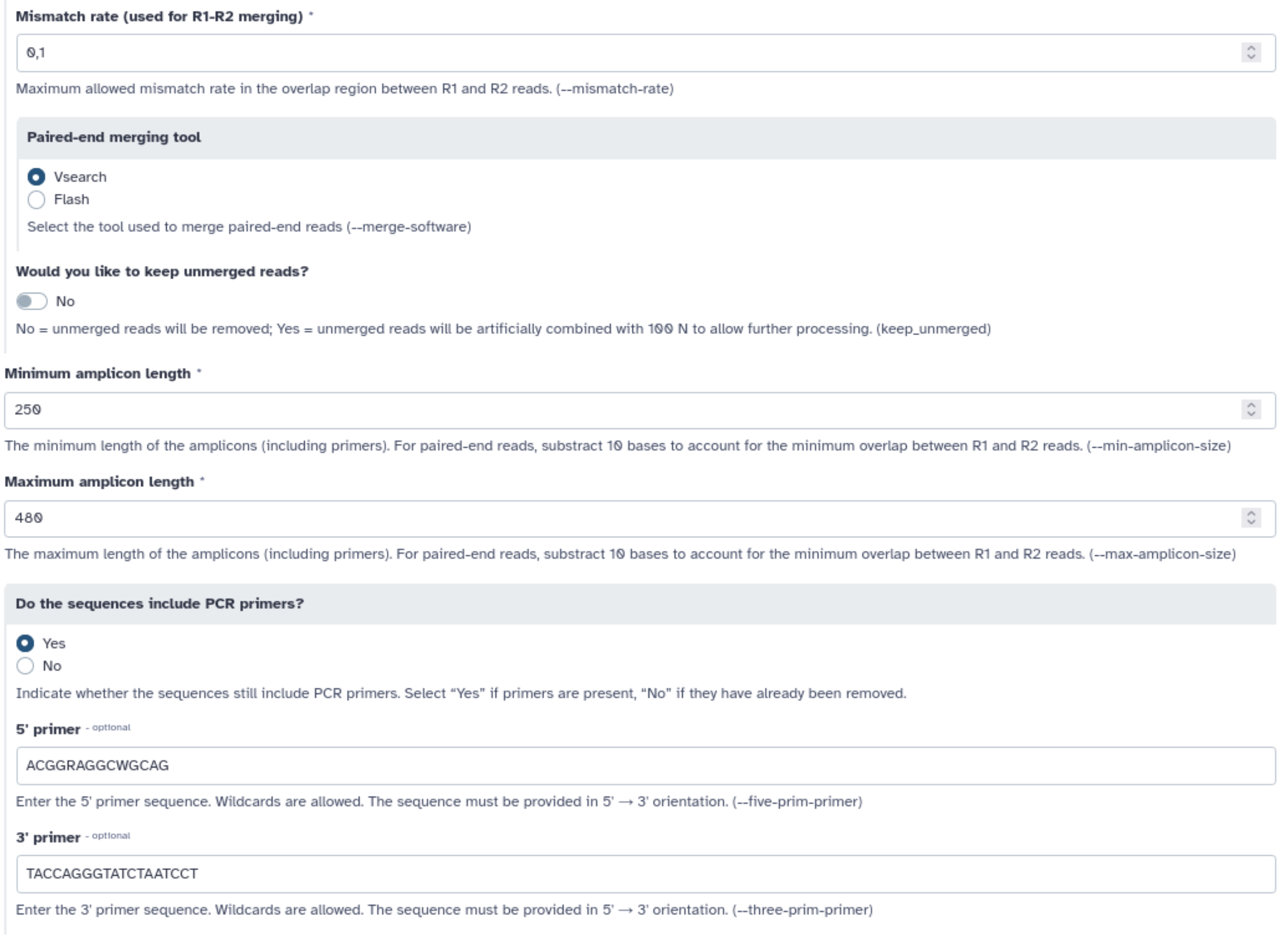
- Read lengths: 250
- Minimum amplicon length: 250
- Maximum amplicon length: 480
- 5’ PCR primer: 5’ ACGGRAGGCWGCAG 3’
- 3’ PCR primer: 3’ TACCAGGGTATCTAATCCT 5’
Vous avez à dispostion un fichier de métadonnées Table 1 (http://genome.jouy.inra.fr/~orue/FROGS_2026/MPC_complete_metadata.tsv) :
et une archive contenant :
- les fichiers 16S (http://genome.jouy.inra.fr/~orue/FROGS_2026/MPC_16S.tar.gz)
- les fichiers ITS (http://genome.jouy.inra.fr/~orue/FROGS_2026/MPC_ITS.tar.gz)
4.1 FROGS Read processing
Supprimez (définitivement) les fichiers FASTQ de l’historique et uploadez l’archive contenant les données 16S
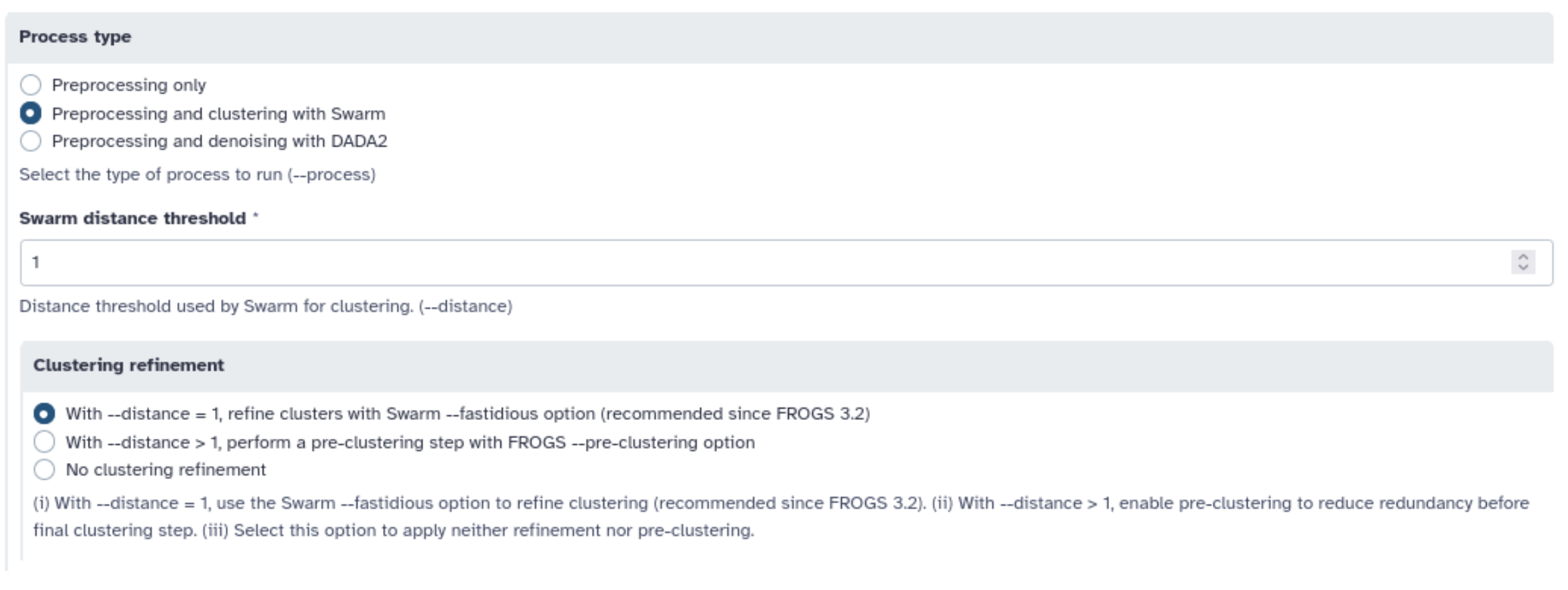
Lancez FROGS reads processing. Utilisez les informations que nous vous avons fournies pour déterminer quels paramètres utiliser
NoteSolution
Avertissement
- L’archive doit être au format
tarsinon elle ne sera pas vue par l’outil ! - Attention au sens de lecture du primer 3’ ! Pour reverse-complémenter un primer : https://reverse-complement.com/
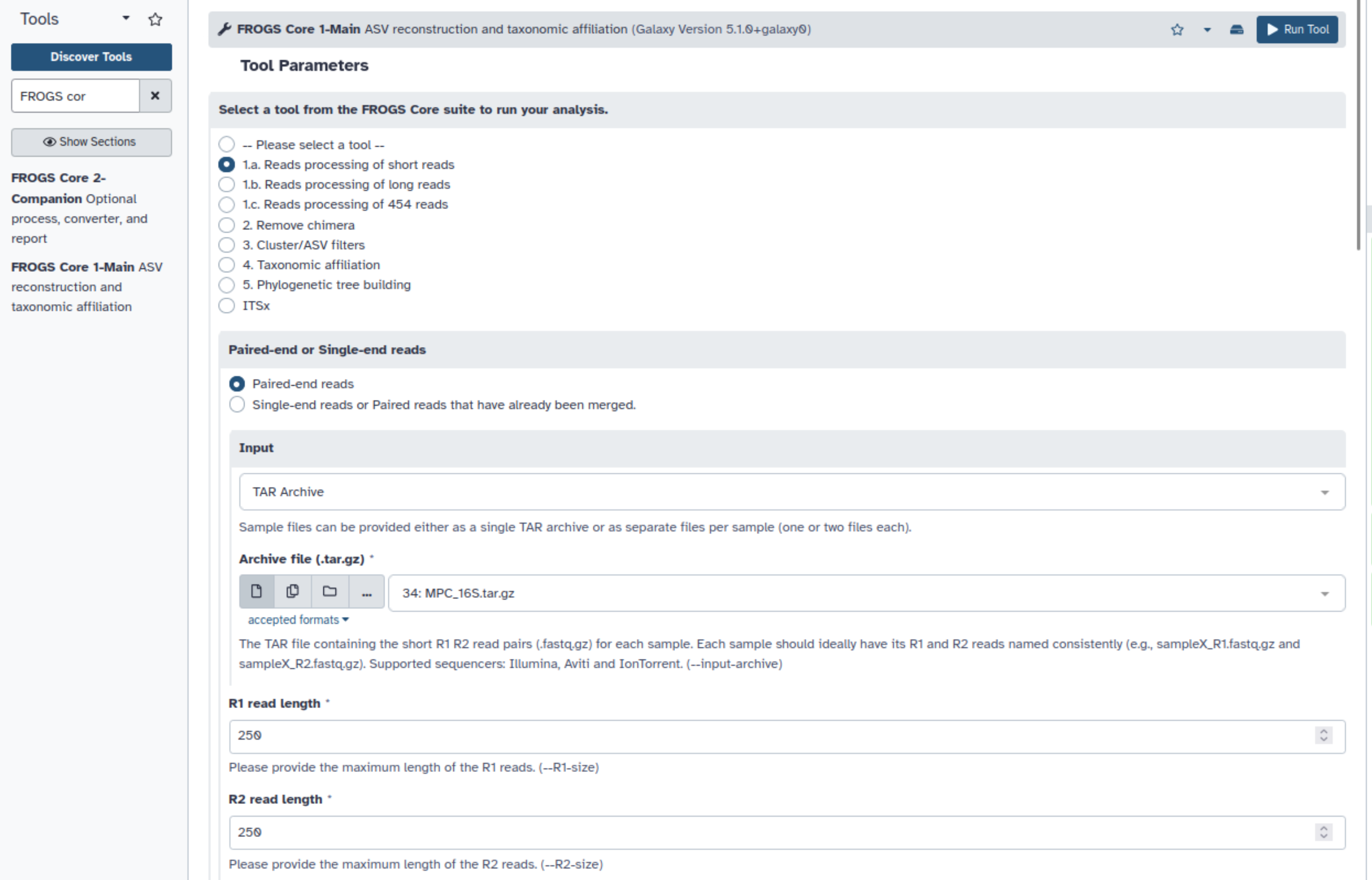
Explorez le rapport HTML généré
Quelle taille d’amplicons choisir ?
NoteSolution
Quand vous ne savez pas, mettez des bornes basses et hautes puis allez regarder la distribution des séquences (bouton Display amplicon lengths) pour définir des bornes plus précises.
Lisez la documentation de l’outil (bas du formulaire) pour comprendre ce qui a été fait
Quelles informations sont présentes dans le fichier FASTA ?
NoteSolution
Dans le fichier FASTA sont stockées les séquences des amplicons
Exemple:
ID_1 TAGGGAATCTTCGGCAATGGGGGCAACCCTGACCGAGCAACGCCGCGTGAGTGAAGAAGGTTTTCGGATCGTAAAGCTCTGTTGTAAGTCAAGAACGGGTGTGAGAGTGGAAAGTTCACACTGTGACGGTAGCTTACCAGAAAGGGACGGCTAACTACGTGCCAGCAGCCGCGGTAATACGTAGGTCCCGAGCGTTGTCCGGATTTATTGGGCGTAAAGCGAGCGCAGGCGGTTTGATAAGTCTGAAGTTAAAGGCTGTGGCTCAACCATAGTTCGC ID_2 TAGGGAATCTTCCACAATGGACGCAAGTCTGATGGAGCAACGCCGCGTGAGTGAAGAAGGTTTTCGGATCGTAAAGCTCTGTTGTTGGTGAAGAAGGATAGAGGCAGTAACTGGTCTTTATTTGACGGTAATCAACCAGAAAGTCACGGCTAACTACGTGCCAGCAGCCGCGGTAATACGTAGGTGGCAAGCGTTGTCCGGATTTATTGGGCGTAAAGCGAGCGCAGGCGGAATGATAAGTCTGATGTGAAAGCCCACGGCTCAACCGTGGAACTG
- Quelles informations sont présentes dans le fichier swarms_composition.txt ?
NoteSolution
Ce fichier est la table de constitution des clusters, une ligne par cluster, et un ou plusieurs identifiants de reads par cluster sur une même ligne
- Est-ce attendu que certaines paires de reads ne contiguent pas ?
NoteSolution
Tout dépend de la taille attendue de l’amplicon qui a été séquencé. Généralement, les régions du 16S ciblées sont censées permettre un chevauchement des paires. Dans ce cas précis, les paires qui ne contiguent pas ne sont pas attendues et peuvent (doivent) être supprimées. Pour les ITS, il est essentiel de les conserver par contre car certains ITS peuvent dépasser 600 paires de bases. Si la taille attendue de votre amplicon est supérieure à 600 bp, vous ne deviez pas avoir de paires qui contiguent.
- Qu’est-ce que la déréplication ?
NoteSolution
La déréplication permet de garder un unique exemplaire de plusieurs séquences identiques. Le détail de l’abondance de cette séquence par échantillon est indiquée dans le fichier count.tsv
- Combien de séquences sont contenues dans le plus gros cluster ?
NoteSolution
137,399
- Combien de clusters sont composés d’une seule séquence ?
NoteSolution
171,973
- Quel pourcentage du total des clusters représentent-ils ?
NoteSolution
95.80 %


4.2 FROGS Remove chimera
- Lancez l’outil FROGS remove chimera pour supprimer les éventuelles chimères que pourrait détecter vsearch [4].
NoteSolution
Explorez le rapport HTML généré
Combien de clusters ont été supprimés ?
NoteSolution
41,976 (23.38%)
- Combien cela représente-t-il de séquences ?
NoteSolution
97,723 (8.32%)
- Qu’en concluez-vous ?
NoteSolution
Les clusters dont la séquence représentative est détectée comme chimérique sont majoritairement composés de peu de séquences.
- Quelle est la plus grande abondance d’un cluster chimérique détecté ?
NoteSolution
157
4.3 FROGS Cluster/ASV filters
- Lancez l’outil FROGS Cluster/ASV filters pour ne garder que les clusters avec une abondance supérieure à 0.005% et présents dans au moins 3 échantillons. Utilisez également la détection des séquences phiX
NoteSolution


Explorez le rapport HTML généré
Combien de clusters ont été supprimés et combien représentent-ils de séquences ?
NoteSolution
99.71% des clusters sont supprimés ! Mais cela représente seulement 14.68% des séquences.
- Quelle information est présente dans le fichier excluded.tsv ?
NoteSolution
Chaque colonne représente un filtre et chaque ligne un cluster. Un cluster peut bien entendu se retrouver dans plusieurs colonnes.
- Quel(s) est (sont) le(s) filtre(s) qui a (ont) permis de supprimer le plus de clusters ?
NoteSolution
La grande majorité des clusters supprimés sont supprimés à la fois parce qu’ils sont vus dans moins de 3 échantillons et avec une abondance relative < 0.00005.
- Ces échantillons étaient-ils contaminés par des séquences de phiX restantes ?
NoteSolution
Non
AstuceAppliquer les filtres suivant le design expérimental
- Utilisez l’information concernant les échantillons répliqués pour ne garder que les clusters présents dans au moins 50% d’un groupe. Le fichier est disponible ici : https://genoweb.toulouse.inra.fr/~formation/15_FROGS/current/chaillou_replicate_information.tsv
Voici à quoi ressemble le fichier
Voilà la section à renseigner :
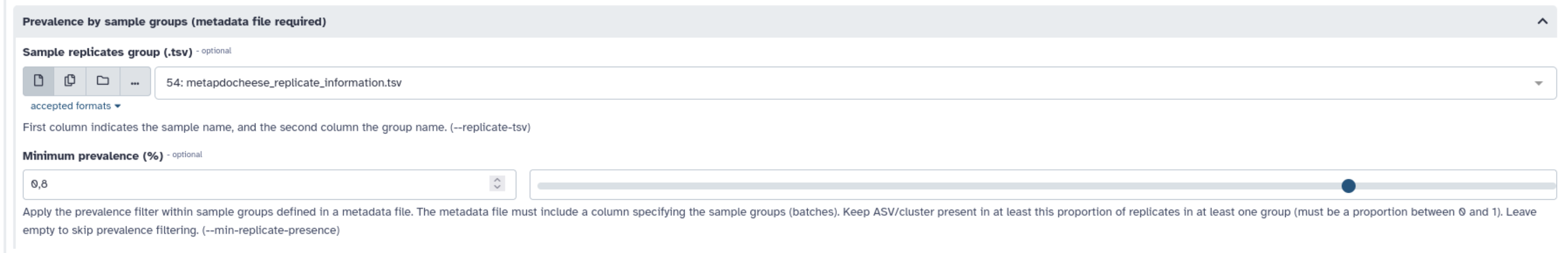
Et la catégorie apparaît dans les filtres appliqués :

4.4 FROGS Taxonomic affiliation
- Lancez l’outil FROGS taxonomic affiliation pour effectuer une affiliation Blast ET RDP avec la base de données DAIRYdb v3.0.0_202304
NoteSolution
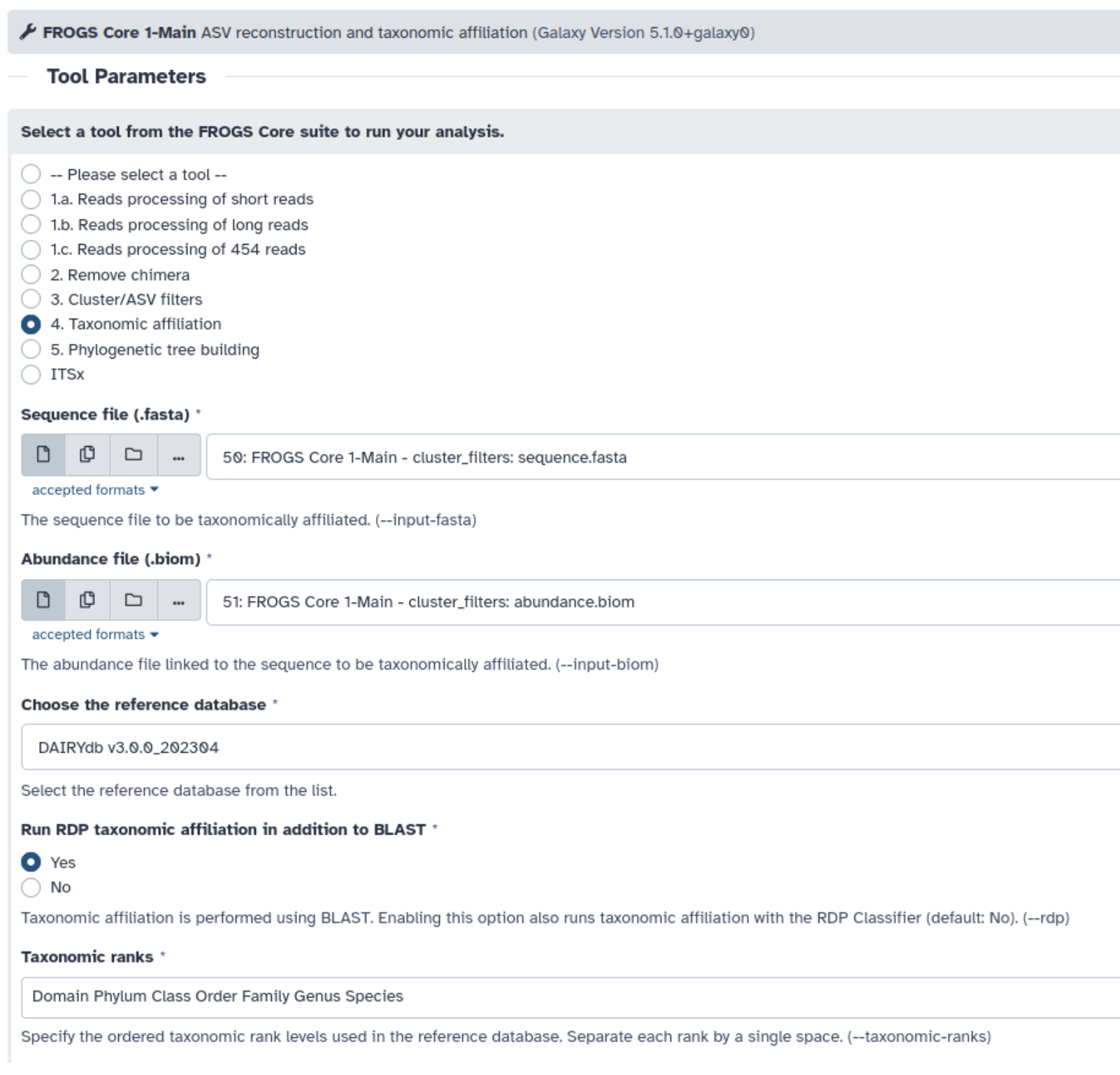
Explorez le rapport HTML généré
Est-ce que tous les ASVs ont été affiliés à une séquence présente dans la base de référence utilisée ?
NoteSolution
Non, 5 ASVs n’ont pas été affiliés. Leur origine est donc à déterminer.
- Que feriez-vous des ASVs qui ne seraient pas affiliés ?
NoteSolution
Il ne faut surtout pas les supprimer. Il s’agit peut-être de séquences non présentes dans la base de données utilisée. Dans ce cas, il faudrait utiliser d’autres bases de données (NCBI par exemple). Il est important de savoir ce qu’elles sont. Elles peuvent être une contamination (hôte, environnement, amplification d’autres espèces…). Cela donnera des informations importantes sur la qualité de votre expérimentation.
- Expliquez le graphique
Blast multi-affiliation summary
NoteSolution
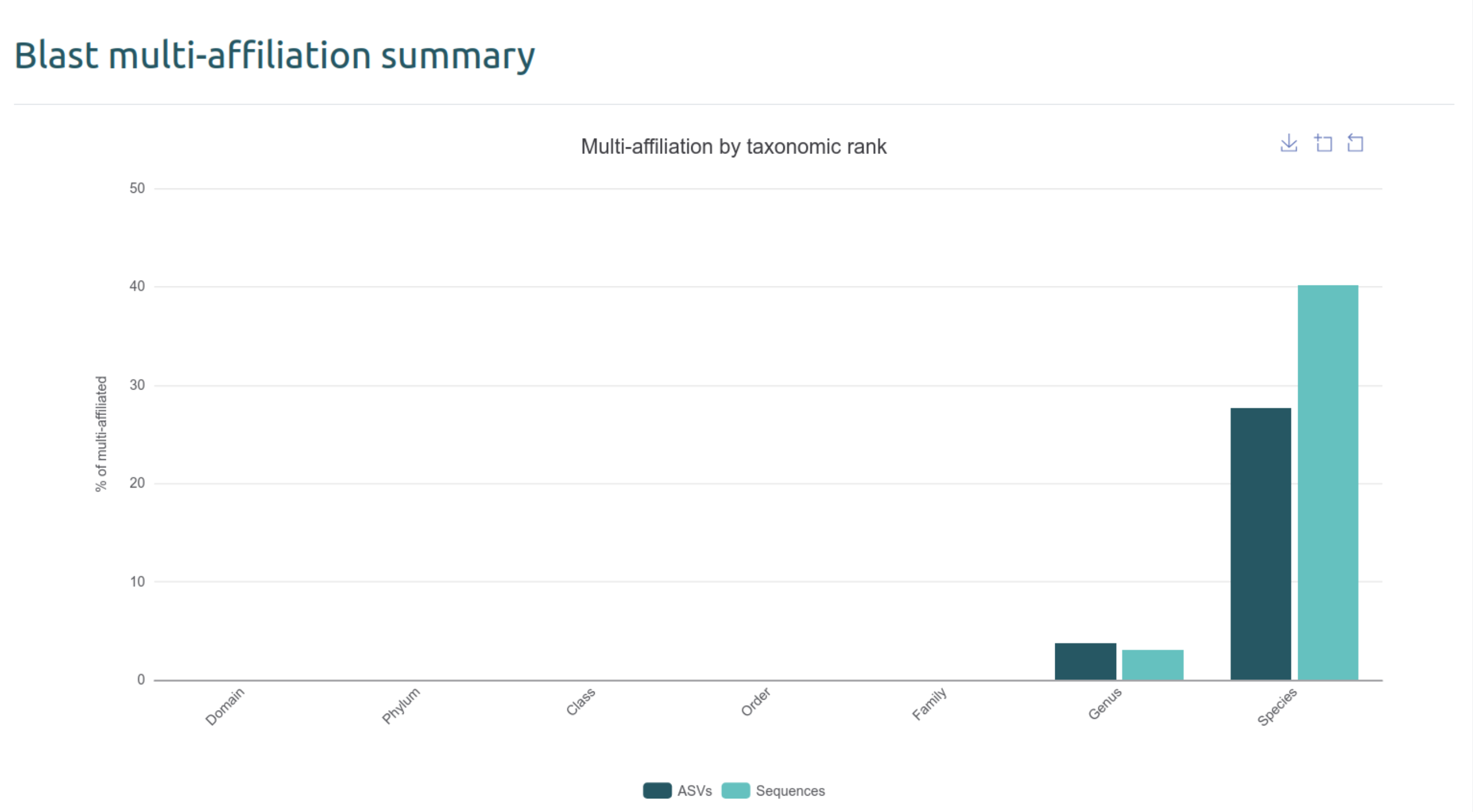
Ce graphique représente le nombre d’ASVs / séquences multi-affiliées à un rang donné. Cela signifie que plusieurs hits identiques de l’ASV sont ressortis avec une ambiguité au rang suivant. Par exemple, pour une multi-affiliation au rang Genus, il y a deux hits d’espèces différentes.
- Le nombre de multi-affiliations au rang
Speciesvous paraît-il surprenant ? Pourquoi ?
NoteSolution
Non, le 16S ne permet généralement pas de discriminer les espèces au-delà du rang Genus.
- Lancez l’outil FROGS affiliation stats
NoteSolution
Explorez le rapport HTML généré
Qu’est-ce qu’une courbe de raréfaction ? Quel est son intérêt principal ?
NoteSolution
Une courbe de raréfaction permet d’évaluer si la profondeur de séquençage est suffisante. Cela permet de dire qu’on a suffisamment séquencé pour que la majorité des espèces de l’échantillon soient séquencées. Il faut dans l’idéal que la courbe atteigne un plateau.
- À quoi vous ferait penser un ASV affilié avec un % de couverture inférieur à 80% ?
NoteSolution
À une potentielle chimère !
- Que conclure si les % d’identité de vos ASVs les plus abondants sont faibles ?
NoteSolution
Vos séquences d’intérêt pourraient ne pas être présentes dans la base de données utilisée. Un petit tour sur LEAP peut vous aider à conclure.
4.5 FROGS Biom to TSV
- Lancez l’outil FROGS biom to tsv en demandant l’extraction des mulit-affiliations et la présence des séquences dans le fichier de sortie
NoteSolution
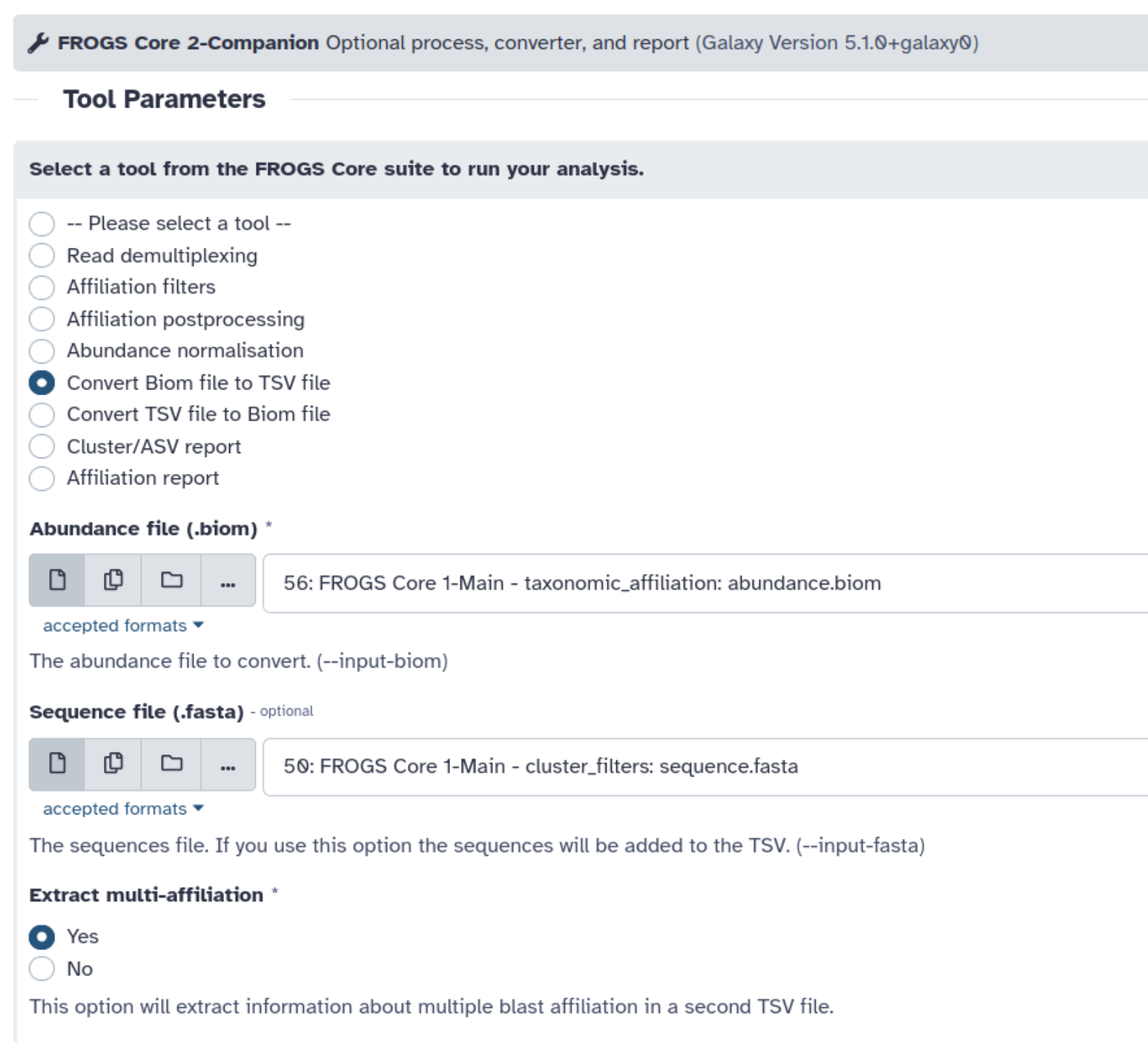
Explorez les datasets générés
Combien de séquences possède le plus gros ASV ?
NoteSolution
137,399 séquences
- Explorez les 5 premiers ASVs et discutez leur affiliation RDP vs. Blast
NoteSolution
Le Cluster_1 est affilié exclusivement à Brochotrix thermosphacta. Vous vous rappelez les copies de 16S dans les génomes bactériens ? Il n’y pas d’ambiguité sur l’espèce, donc pas de multi-affiliation ici.
| ASV | Explication |
|---|---|
| ID_1 | Après avoir regardé les multi-affiliations, il devrait bien s’agir de Corynebacterium_variabile comme le dit RDP. |
| ID_2 | Pas de doute : Streptococcus_thermophilus |
| ID_3 | RDP me dit Lactococcus_lactis. Pourtant, les multi-affiliations me disent Lactococcus_cremoris ou Lactococcus_lactis avec 100% d’identité. Donc un 16S identique. On ne peut pas savoir !!! On laisse comme ça |
| ID_4 | Lactobacillus_delbrueckii, aucun doute |
| ID_5 | Halomonas_variabilis, aucun doute |
Pour vous faciliter la vie, nous avons développé une interface web : AffiliationExplorer qui vous permet de modifier certaines multi-affiliations si nécessaire. Pour cela, il vous faudra télécharger sur votre ordinateur le fichier BIOM et le fichier de multi-affiliations, ainsi que le FASTA si vous avez éventuellement besoin de la séquence pour faire vos modifications.
https://shiny.migale.inrae.fr/app_direct/affiliationexplorer/
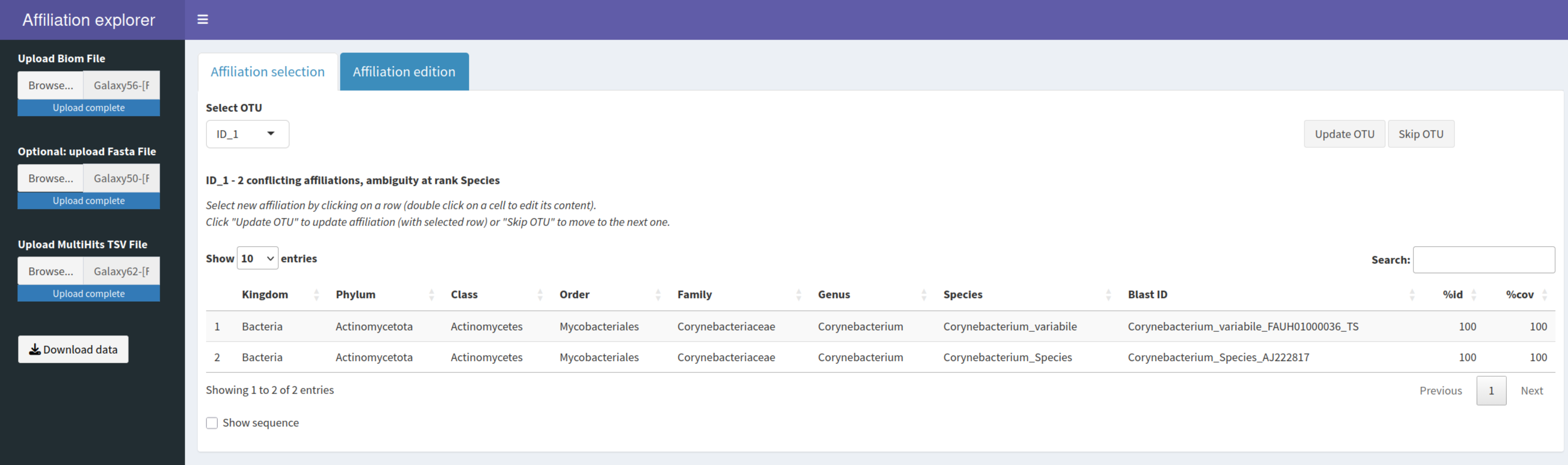
NoteDemonstration pour modifier une affiliation existante
4.6 FROGS Tree
- Lancez l’outil FROGS tree
NoteSolution
Explorez le rapport HTML généré
Que faire si certains ASVs ne sont pas inclus dans l’arbre ?
NoteSolution
Ceux-là seront inexploitables pour calculer des indices basés sur la distance phylogénétique.
5 Toy datasets
5.1 16S
- Data from https://doi.org/10.1371/journal.pone.0204629
- http://genome.jouy.inra.fr/~orue/data.tar.gz
- 16S V3-V4
- V3F(5’-ACGGRAGGCWGCAGT-3’) and V4R (5’-TACCAGGGTATCTAATCCT-3’)
6 Références
1. Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, et al. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics. 2018;34:1287‑94. doi:10.1093/bioinformatics/btx791.
2. Andrews S. FastQC A Quality Control tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
3. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047‑8.
4. Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
