library(edgeR) # analysis of digital gene expression
library(kableExtra) # construct complex table
library(magrittr) # provides the pipe operator
library(dplyr) # grammar of data manipulation
library(ggplot2) # data visualisation
library(stringr) # string operators
library(ViSEAGO) # enrichment testRNA-seq transcriptome analysis
Study of chorioallantoic membrane of chicken embryos
RNA-Seq
R
Introduction
This vignette supports the differential analysis and the functional enrichment test of the dataset described in the data paper entitled “RNA-seq dataset of the chorioallantoic membrane of males and females chicken embryos, after 11 and 15 days of incubation” [1].
Setup
R Packages installed for these analyses.
Dataset
This dataset contains the gene expression of chorioallantoic membrane (CAM) of male and female chicken embryos, after 11 and 15 days of incubation. To easy manipulate data, a DGEList objectwas constructed. It contains a matrix counts, a data.frame samples describing the samples and the informations about sequencing, and a data.frame genes containing annotation for the genes obtained using biomaRt
# read dge object
dge <- readRDS("./cam-data/cam_dge_tuto.rds")
dgeAn object of class "DGEList"
$samples
files group lib.size norm.factors labels stade
1 Readcount_Ech_C11_F_11.txt EID11F 34020303 1 EID11F_11 EID11
2 Readcount_Ech_C11_F_20.txt EID11F 39748001 1 EID11F_20 EID11
3 Readcount_Ech_C11_F_22.txt EID11F 40176549 1 EID11F_22 EID11
4 Readcount_Ech_C11_F_26.txt EID11F 34417922 1 EID11F_26 EID11
5 Readcount_Ech_C11_F_29.txt EID11F 40019118 1 EID11F_29 EID11
sex num_sample
1 Female 11
2 Female 20
3 Female 22
4 Female 26
5 Female 29
34 more rows ...
$counts
Samples
Tags C11_F_11 C11_F_20 C11_F_22 C11_F_26 C11_F_29 C11_F_3
ENSGALG00000000011 1004 1212 982 967 1166 795
ENSGALG00000000038 0 0 0 0 0 9
ENSGALG00000000044 7 32 114 71 165 9
ENSGALG00000000048 6430 5351 8774 7411 10834 4672
ENSGALG00000000055 3531 3457 3277 2889 3506 2353
Samples
Tags C11_F_39 C11_F_41 C11_F_46 C11_F_6 C11_M_14 C11_M_16
ENSGALG00000000011 795 886 947 1137 914 973
ENSGALG00000000038 0 5 0 6 5 0
ENSGALG00000000044 15 53 70 60 33 22
ENSGALG00000000048 5425 11509 6604 3627 5262 5040
ENSGALG00000000055 3067 3042 2821 3230 2959 2649
Samples
Tags C11_M_2 C11_M_24 C11_M_27 C11_M_38 C11_M_44 C11_M_48
ENSGALG00000000011 1033 947 959 1207 1097 997
ENSGALG00000000038 0 1 1 0 6 1
ENSGALG00000000044 23 100 106 177 272 62
ENSGALG00000000048 4823 7309 4772 8812 9016 9904
ENSGALG00000000055 2910 2954 2971 3256 3318 2671
Samples
Tags C11_M_7 C11_M_9 C15_F_102 C15_F_54 C15_F_59 C15_F_64
ENSGALG00000000011 852 1182 871 881 1135 1336
ENSGALG00000000038 0 0 1 1 1 1
ENSGALG00000000044 11 35 85 19 3 3
ENSGALG00000000048 5887 3631 29829 23884 45168 55303
ENSGALG00000000055 2525 3296 2796 2941 3664 4334
Samples
Tags C15_F_70 C15_F_74 C15_F_79 C15_F_88 C15_F_95 C15_M_100
ENSGALG00000000011 869 825 844 679 960 789
ENSGALG00000000038 0 0 0 2 1 0
ENSGALG00000000044 30 15 6 1 9 13
ENSGALG00000000048 26385 30697 33933 30393 56321 38938
ENSGALG00000000055 3107 2948 2760 3033 3959 3010
Samples
Tags C15_M_52 C15_M_57 C15_M_67 C15_M_72 C15_M_76 C15_M_81
ENSGALG00000000011 939 775 1113 817 786 936
ENSGALG00000000038 0 7 1 0 0 2
ENSGALG00000000044 4 15 16 6 6 10
ENSGALG00000000048 44870 13048 25992 20599 33184 46262
ENSGALG00000000055 3169 2961 3151 2878 3428 4027
Samples
Tags C15_M_86 C15_M_93 C15_M_98
ENSGALG00000000011 860 846 782
ENSGALG00000000038 1 5 4
ENSGALG00000000044 12 4 9
ENSGALG00000000048 32289 38850 44653
ENSGALG00000000055 2986 2871 3092
26146 more rows ...
$genes
gene_id external_gene_name
1 ENSGALG00000000011 C10orf88
2 ENSGALG00000000038 CTRB2
3 ENSGALG00000000044 WFIKKN1
4 ENSGALG00000000048
5 ENSGALG00000000055 LAMTOR3
description
1 chromosome 10 open reading frame 88 [Source:HGNC Symbol;Acc:HGNC:25822]
2 chymotrypsinogen B2 [Source:NCBI gene;Acc:431235]
3 WAP, follistatin/kazal, immunoglobulin, kunitz and netrin domain containing 1 [Source:HGNC Symbol;Acc:HGNC:30912]
4 V-type proton ATPase catalytic subunit A-like [Source:NCBI gene;Acc:776719]
5 late endosomal/lysosomal adaptor, MAPK and MTOR activator 3 [Source:NCBI gene;Acc:425210]
hgnc_symbol chromosome_name
1 C10orf88 6
2 11
3 WFIKKN1 14
4 25
5 4
26146 more rows ...# convert variables into factors
dge$samples <- dge$samples %>%
mutate(group = as.factor(group),
sex = as.factor(sex),
stade = as.factor(stade))In this study, the effect of stade with (EID11, EID15) levels and sex with (Female, Male) levels will be explore. The number of biological repetitions is 10, 10, 9, 10 for EID11F, EID11M, EID15F, EID15M, respectively. As mentioned in the paper, the sample numbered 83 (EID15 female) was removed.
The data.frame samples contains a column lib.size for the library size or sequencing depth for each sample, equal to the sum of the aligned reads.
Differential analysis
Statistical analyses were performed using edgeR
Genes with very low counts across all libraries may be filtered.
# Filter gene with low count, at least number of biological replicates with cpm > valfilt
valfilt <- 1
nbrepbio <- min(table(dge$samples$group))
keep <- rowSums(edgeR::cpm(dge) > valfilt) >= nbrepbio
dge <- dge[keep, , keep.lib.sizes = FALSE]
dim(dge)[1] 15124 39The filtering on genes is based on count per million (cpm) greater than 1 in at least 9 samples corresponding to the minimum number of biological replicates. We kept 15124 expressed genes for further analyses from 39 samples.
Normalisation
A normalization factor is calculated to take into account the different sizes of the sequencing banks (i.e. the total read count) and the distribution of reads per sample on sequencing run, as discussed [3]. Normalization by trimmed mean of M values (TMM) [2] is performed by using the calcNormFactors function from edgeR
Here, the table contains library size and normalisation factors using TMM method ordered by library size. There are also some useful graphs to support the quality control performed during the sequencing steps and bioinformatics analyses.
dge <- calcNormFactors(dge)
dge$samples[order(dge$samples$lib.size), ] %>%
kbl() %>%
kable_styling() %>%
scroll_box(width = "100%", height = "200px")| files | group | lib.size | norm.factors | labels | stade | sex | num_sample | |
|---|---|---|---|---|---|---|---|---|
| 6 | Readcount_Ech_C11_F_3.txt | EID11F | 28239877 | 0.9707889 | EID11F_3 | EID11 | Female | 3 |
| 12 | Readcount_Ech_C11_M_16.txt | EID11M | 30753266 | 0.9678185 | EID11M_16 | EID11 | Male | 16 |
| 27 | Readcount_Ech_C15_F_79.txt | EID15F | 30829131 | 1.0158022 | EID15F_79 | EID15 | Female | 79 |
| 7 | Readcount_Ech_C11_F_39.txt | EID11F | 30988543 | 1.0123821 | EID11F_39 | EID11 | Female | 39 |
| 19 | Readcount_Ech_C11_M_7.txt | EID11M | 31208638 | 0.9970126 | EID11M_7 | EID11 | Male | 7 |
| 21 | Readcount_Ech_C15_F_102.txt | EID15F | 32221501 | 0.9802677 | EID15F_102 | EID15 | Female | 102 |
| 34 | Readcount_Ech_C15_M_72.txt | EID15M | 32432332 | 1.0073170 | EID15M_72 | EID15 | Male | 72 |
| 11 | Readcount_Ech_C11_M_14.txt | EID11M | 32899106 | 0.9673810 | EID11M_14 | EID11 | Male | 14 |
| 9 | Readcount_Ech_C11_F_46.txt | EID11F | 33353072 | 1.0178110 | EID11F_46 | EID11 | Female | 46 |
| 8 | Readcount_Ech_C11_F_41.txt | EID11F | 33622506 | 1.0429180 | EID11F_41 | EID11 | Female | 41 |
| 1 | Readcount_Ech_C11_F_11.txt | EID11F | 33981408 | 0.9898948 | EID11F_11 | EID11 | Female | 11 |
| 37 | Readcount_Ech_C15_M_86.txt | EID15M | 34166458 | 1.0071100 | EID15M_86 | EID15 | Male | 86 |
| 39 | Readcount_Ech_C15_M_98.txt | EID15M | 34220625 | 0.9887918 | EID15M_98 | EID15 | Male | 98 |
| 30 | Readcount_Ech_C15_M_100.txt | EID15M | 34249920 | 1.0060331 | EID15M_100 | EID15 | Male | 100 |
| 38 | Readcount_Ech_C15_M_93.txt | EID15M | 34270870 | 0.9919875 | EID15M_93 | EID15 | Male | 93 |
| 4 | Readcount_Ech_C11_F_26.txt | EID11F | 34369303 | 1.0599808 | EID11F_26 | EID11 | Female | 26 |
| 22 | Readcount_Ech_C15_F_54.txt | EID15F | 34403366 | 1.0024428 | EID15F_54 | EID15 | Female | 54 |
| 28 | Readcount_Ech_C15_F_88.txt | EID15F | 34515320 | 1.0000987 | EID15F_88 | EID15 | Female | 88 |
| 15 | Readcount_Ech_C11_M_27.txt | EID11M | 34704465 | 1.0311382 | EID11M_27 | EID11 | Male | 27 |
| 18 | Readcount_Ech_C11_M_48.txt | EID11M | 35116336 | 1.0623694 | EID11M_48 | EID11 | Male | 48 |
| 26 | Readcount_Ech_C15_F_74.txt | EID15F | 35566461 | 0.9961322 | EID15F_74 | EID15 | Female | 74 |
| 31 | Readcount_Ech_C15_M_52.txt | EID15M | 35666960 | 0.9866407 | EID15M_52 | EID15 | Male | 52 |
| 25 | Readcount_Ech_C15_F_70.txt | EID15F | 35711970 | 1.0172644 | EID15F_70 | EID15 | Female | 70 |
| 13 | Readcount_Ech_C11_M_2.txt | EID11M | 35760262 | 0.9711445 | EID11M_2 | EID11 | Male | 2 |
| 20 | Readcount_Ech_C11_M_9.txt | EID11M | 36244024 | 0.9700712 | EID11M_9 | EID11 | Male | 9 |
| 35 | Readcount_Ech_C15_M_76.txt | EID15M | 36423127 | 1.0053627 | EID15M_76 | EID15 | Male | 76 |
| 10 | Readcount_Ech_C11_F_6.txt | EID11F | 36634581 | 0.9163450 | EID11F_6 | EID11 | Female | 6 |
| 32 | Readcount_Ech_C15_M_57.txt | EID15M | 36729326 | 1.0033506 | EID15M_57 | EID15 | Male | 57 |
| 14 | Readcount_Ech_C11_M_24.txt | EID11M | 38256961 | 1.0244010 | EID11M_24 | EID11 | Male | 24 |
| 33 | Readcount_Ech_C15_M_67.txt | EID15M | 38400121 | 0.9927465 | EID15M_67 | EID15 | Male | 67 |
| 36 | Readcount_Ech_C15_M_81.txt | EID15M | 38612243 | 0.9411493 | EID15M_81 | EID15 | Male | 81 |
| 16 | Readcount_Ech_C11_M_38.txt | EID11M | 39030485 | 1.0348989 | EID11M_38 | EID11 | Male | 38 |
| 17 | Readcount_Ech_C11_M_44.txt | EID11M | 39274952 | 1.0553474 | EID11M_44 | EID11 | Male | 44 |
| 2 | Readcount_Ech_C11_F_20.txt | EID11F | 39700690 | 0.9793198 | EID11F_20 | EID11 | Female | 20 |
| 5 | Readcount_Ech_C11_F_29.txt | EID11F | 39971511 | 1.0200657 | EID11F_29 | EID11 | Female | 29 |
| 3 | Readcount_Ech_C11_F_22.txt | EID11F | 40117899 | 1.0346771 | EID11F_22 | EID11 | Female | 22 |
| 23 | Readcount_Ech_C15_F_59.txt | EID15F | 40729955 | 0.9517567 | EID15F_59 | EID15 | Female | 59 |
| 29 | Readcount_Ech_C15_F_95.txt | EID15F | 43711444 | 0.9940782 | EID15F_95 | EID15 | Female | 95 |
| 24 | Readcount_Ech_C15_F_64.txt | EID15F | 49181211 | 1.0044379 | EID15F_64 | EID15 | Female | 64 |
# barplot of scaling factors
p<-ggplot(dge$samples, aes(x=dge$samples$labels, y=dge$samples$norm.factors, fill=dge$samples$group)) +
geom_col() + xlab("Samples") + ylab("TMM factors") + labs(fill = "group") + theme_bw() +
theme(axis.text.x = element_text(angle=90)) + ggtitle("TMM size factors \n")
p 
# barplot of library size with group information
p<-ggplot(dge$samples, aes(x=dge$samples$labels, y=dge$samples$lib.size, fill=dge$samples$group)) +
geom_col() + xlab("Samples") + ylab("Library size") + labs(fill = "group") + theme_bw() +
theme(axis.text.x = element_text(angle=90)) + ggtitle("Library size \n")
p 
Multidimensional scaling plot
Multidimensional scaling (MDS) plot shows the relationships between the samples. The top (500 genes) are used to calculate the distance between expression profiles of samples. The distance approximate the log2 fold change between the samples.
As expected, samples are grouped by stade and sex.
limma::plotMDS(dge, col = as.numeric(dge$samples$group), labels = dge$samples$num_sample, cex=0.9, gene.selection = "pairwise", top = 500, main = "MDS plot with top 500 genes")
legend("topleft", legend = levels(dge$samples$group), col = unique(as.numeric(dge$samples$group)),
cex = 0.8, lwd = 1)
Differential analysis
The aim is to identify the genes differentially expressed between sex at each day of incubation (stade variable). At this step, read counts were normalized using trimmed mean of M-values (TMM) method [2] from edgeR
First, the matrix design was computed.
#matrix design: one factor group
design <- model.matrix(~ 0 + group, data = dge$samples)
colnames(design) <- colnames(design) %>% str_remove(., "group")
design EID11F EID11M EID15F EID15M
1 1 0 0 0
2 1 0 0 0
3 1 0 0 0
4 1 0 0 0
5 1 0 0 0
6 1 0 0 0
7 1 0 0 0
8 1 0 0 0
9 1 0 0 0
10 1 0 0 0
11 0 1 0 0
12 0 1 0 0
13 0 1 0 0
14 0 1 0 0
15 0 1 0 0
16 0 1 0 0
17 0 1 0 0
18 0 1 0 0
19 0 1 0 0
20 0 1 0 0
21 0 0 1 0
22 0 0 1 0
23 0 0 1 0
24 0 0 1 0
25 0 0 1 0
26 0 0 1 0
27 0 0 1 0
28 0 0 1 0
29 0 0 1 0
30 0 0 0 1
31 0 0 0 1
32 0 0 0 1
33 0 0 0 1
34 0 0 0 1
35 0 0 0 1
36 0 0 0 1
37 0 0 0 1
38 0 0 0 1
39 0 0 0 1
attr(,"assign")
[1] 1 1 1 1
attr(,"contrasts")
attr(,"contrasts")$group
[1] "contr.treatment"Genes differentially expressed between CAM males and females were obtained for each incubation day using the likelihood ratio test on the two defined contrasts EID11_MvsF = EID11M - EID11F and EID15_MvsF = EID15M - EID15F.
# differences between sex (M versus F) within EID11
EID11_MvsF <- makeContrasts(EID11M - EID11F, levels=colnames(design))
# differences between sex (M versus F) within EID15
EID15_MvsF <- makeContrasts(EID15M - EID15F, levels=colnames(design))
my.contrasts <- list(EID11_MvsF = EID11_MvsF,
EID15_MvsF = EID15_MvsF)We set robust=TRUE when estimating the dispersion. This has no effect on the downstream analysis, but is nevertheless very useful for identifying outliers from the trend of the mean dispersion of the negative binomial distribution.
# robust at gene level
# with specific design
dge<-estimateDisp(dge, design = design, robust=TRUE)
#summary(dge$tagwise.dispersion)
plotBCV(dge)
We used GLM model [4] to perform likelihood ratio tests for each contrast defined as EID11_MvsF, EID15_MvsF. List of differentially expressed genes were obtained after controlling the false positive rate with a Benjamini-Hochberg correction [5] at 0.05 threshold.
# model GLM with the design
fit <- glmFit(dge, design = design)lrt <- glmLRT(fit, contrast = EID11_MvsF)
# Number of DE genes
summary(decideTests(lrt)) -1*EID11F 1*EID11M
Down 157
NotSig 14525
Up 442# Table of DE genes results
res <- topTags(lrt, n=nrow(dge$counts), adjust.method="BH", sort.by="none")$table
# see the first rows
head(res) gene_id external_gene_name
ENSGALG00000000011 ENSGALG00000000011 C10orf88
ENSGALG00000000044 ENSGALG00000000044 WFIKKN1
ENSGALG00000000048 ENSGALG00000000048
ENSGALG00000000055 ENSGALG00000000055 LAMTOR3
ENSGALG00000000059 ENSGALG00000000059 TUBB3
ENSGALG00000000067 ENSGALG00000000067 SPR
description
ENSGALG00000000011 chromosome 10 open reading frame 88 [Source:HGNC Symbol;Acc:HGNC:25822]
ENSGALG00000000044 WAP, follistatin/kazal, immunoglobulin, kunitz and netrin domain containing 1 [Source:HGNC Symbol;Acc:HGNC:30912]
ENSGALG00000000048 V-type proton ATPase catalytic subunit A-like [Source:NCBI gene;Acc:776719]
ENSGALG00000000055 late endosomal/lysosomal adaptor, MAPK and MTOR activator 3 [Source:NCBI gene;Acc:425210]
ENSGALG00000000059 tubulin, beta 3 class III [Source:NCBI gene;Acc:431043]
ENSGALG00000000067 sepiapterin reductase (7,8-dihydrobiopterin:NADP+ oxidoreductase) [Source:NCBI gene;Acc:425255]
hgnc_symbol chromosome_name logFC logCPM LR
ENSGALG00000000011 C10orf88 6 0.02516631 4.7368237 0.1103700
ENSGALG00000000044 WFIKKN1 14 0.45759194 0.3106141 0.7125593
ENSGALG00000000048 25 -0.15043214 9.1528049 0.6450647
ENSGALG00000000055 4 -0.09488628 6.4495919 2.7667930
ENSGALG00000000059 11 0.41117847 7.8279704 2.8421317
ENSGALG00000000067 SPR 4 0.09039913 4.5458272 1.0399541
PValue FDR
ENSGALG00000000011 0.73972326 0.9886434
ENSGALG00000000044 0.39859549 0.9436440
ENSGALG00000000048 0.42188272 0.9544246
ENSGALG00000000055 0.09623880 0.6636528
ENSGALG00000000059 0.09182232 0.6591022
ENSGALG00000000067 0.30783215 0.9078363Histogram of raw p-values and mean-difference plot (also called MA-plot) are useful graphs to validate the model.
# histogram of raw pvalues
p <- ggplot(data = lrt$table, aes(x = PValue))
p <- p + geom_histogram(color="darkblue", fill="lightblue")
p <- p + theme_bw() + xlab("Raw pvalue")
p <- p + ggtitle("Histogram of raw pvalues for EID11_MvsF")
print(p)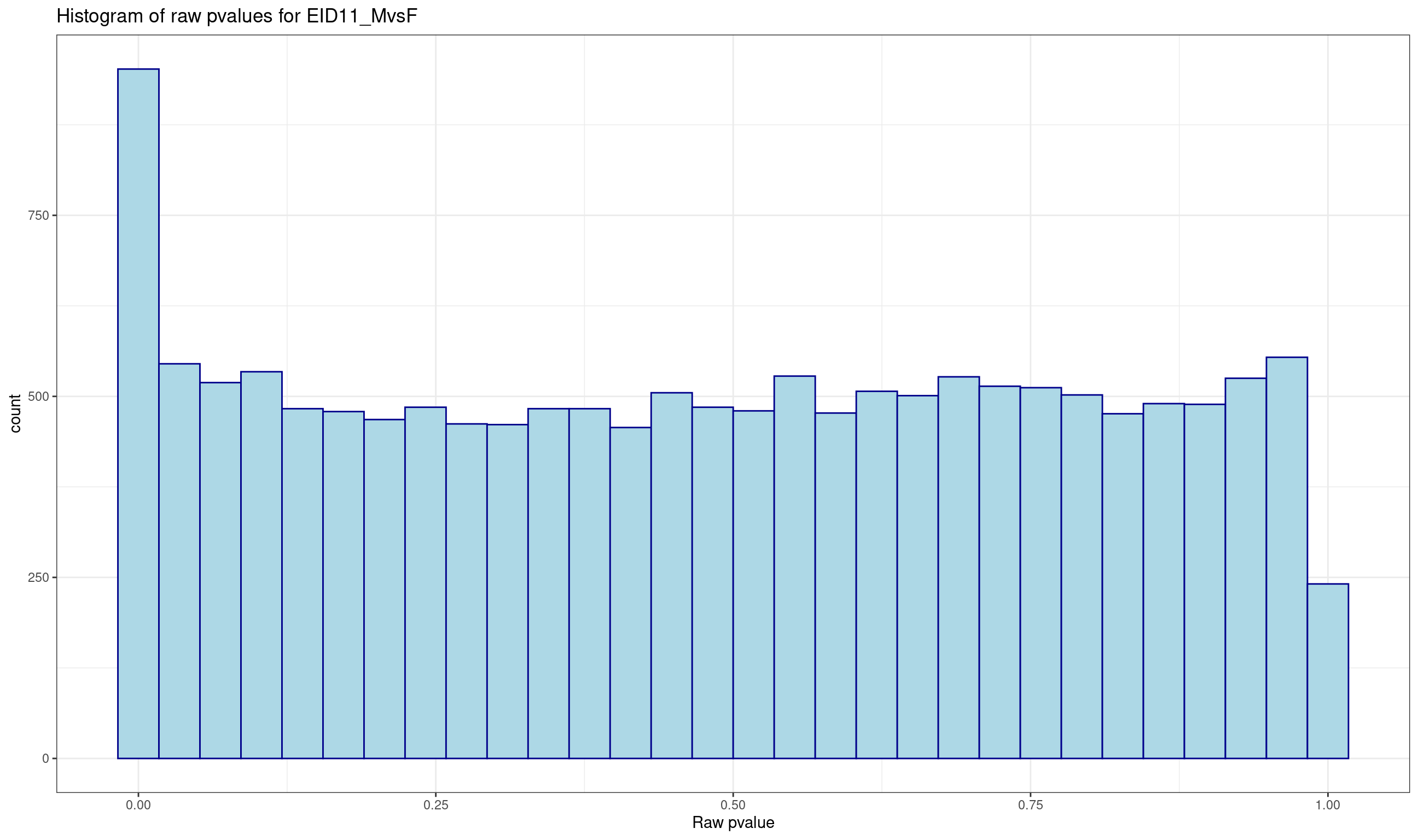
# MA plot
plotMD(lrt, main = "MA plot for EID11_MvsF")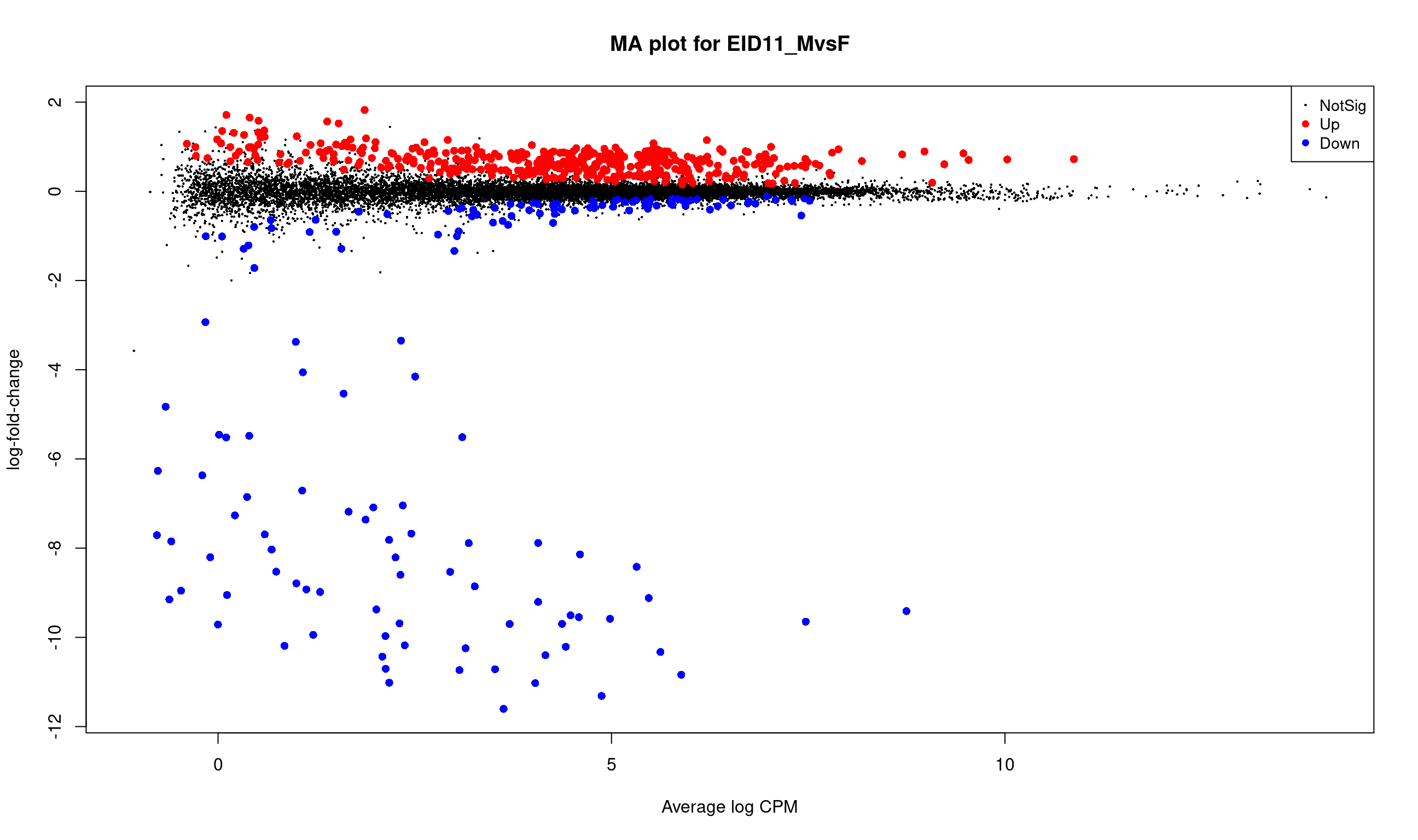
Volcano plots represent a useful way to visualise the results of differential expression analyses. The x-axis is log2 fold change and represents biological significance. The y-axis is -Log10 Pvalue of the contrast tested and represents statistical significance. The highest genes on the graph are the most significantly differentially expressed (DE) genes.
The EnhancedVolcano
# volcano plot
p <- ggplot(data = res, aes(x = logFC, y = -log10(FDR)))
p <- p + geom_point(size=2, shape=19)
p <- p + ggtitle("Volcanoplot: Difference between males and females at EID11")
p
Save the results in separated files using a comma for the decimal point and a semicolon for the separator between columns.
# complete - all genes after filtering step
EID11_MvsF_bg <- res
write.csv2(EID11_MvsF_bg, file="output/complete_EID11_males_vs_females.csv")
# up regulated DE genes at FDR threshold 0.05
EID11_MvsF_up <- dplyr::filter(res, FDR <= 0.05 & logFC > 0)
write.csv2(EID11_MvsF_up, file="output/up_EID11_males_vs_females.csv")
# down regulated DE genes at FDR threshold 0.05
EID11_MvsF_down <- dplyr::filter(res, FDR <= 0.05 & logFC < 0)
write.csv2(EID11_MvsF_down, file="output/down_EID11_males_vs_females.csv")lrt <- glmLRT(fit, contrast = EID15_MvsF)
# Number of DE genes
summary(decideTests(lrt)) -1*EID15F 1*EID15M
Down 134
NotSig 14551
Up 439# Table of DE genes results
res <- topTags(lrt, n=nrow(dge$counts), adjust.method="BH", sort.by="none")$table
# see the first rows
head(res) gene_id external_gene_name
ENSGALG00000000011 ENSGALG00000000011 C10orf88
ENSGALG00000000044 ENSGALG00000000044 WFIKKN1
ENSGALG00000000048 ENSGALG00000000048
ENSGALG00000000055 ENSGALG00000000055 LAMTOR3
ENSGALG00000000059 ENSGALG00000000059 TUBB3
ENSGALG00000000067 ENSGALG00000000067 SPR
description
ENSGALG00000000011 chromosome 10 open reading frame 88 [Source:HGNC Symbol;Acc:HGNC:25822]
ENSGALG00000000044 WAP, follistatin/kazal, immunoglobulin, kunitz and netrin domain containing 1 [Source:HGNC Symbol;Acc:HGNC:30912]
ENSGALG00000000048 V-type proton ATPase catalytic subunit A-like [Source:NCBI gene;Acc:776719]
ENSGALG00000000055 late endosomal/lysosomal adaptor, MAPK and MTOR activator 3 [Source:NCBI gene;Acc:425210]
ENSGALG00000000059 tubulin, beta 3 class III [Source:NCBI gene;Acc:431043]
ENSGALG00000000067 sepiapterin reductase (7,8-dihydrobiopterin:NADP+ oxidoreductase) [Source:NCBI gene;Acc:425255]
hgnc_symbol chromosome_name logFC logCPM
ENSGALG00000000011 C10orf88 6 -0.03122908 4.7368237
ENSGALG00000000044 WFIKKN1 14 -1.05217055 0.3106141
ENSGALG00000000048 25 -0.01456782 9.1528049
ENSGALG00000000055 4 0.02539276 6.4495919
ENSGALG00000000059 11 -0.23793501 7.8279704
ENSGALG00000000067 SPR 4 0.09422590 4.5458272
LR PValue FDR
ENSGALG00000000011 0.159739642 0.68939634 0.9996332
ENSGALG00000000044 3.318413100 0.06850793 0.6500799
ENSGALG00000000048 0.005742689 0.93959370 0.9996332
ENSGALG00000000055 0.188175870 0.66443918 0.9996332
ENSGALG00000000059 0.906407523 0.34106941 0.9996332
ENSGALG00000000067 1.058917896 0.30346212 0.9955664Histogram of raw p-values and mean-difference plot (also called MA-plot) are useful graphs to validate the model.
# histogram of raw pvalues
p <- ggplot(data = lrt$table, aes(x = PValue))
p <- p + geom_histogram(color="darkblue", fill="lightblue")
p <- p + theme_bw() + xlab("Raw pvalue")
p <- p + ggtitle("Histogram of raw pvalues for EID15_MvsF")
print(p)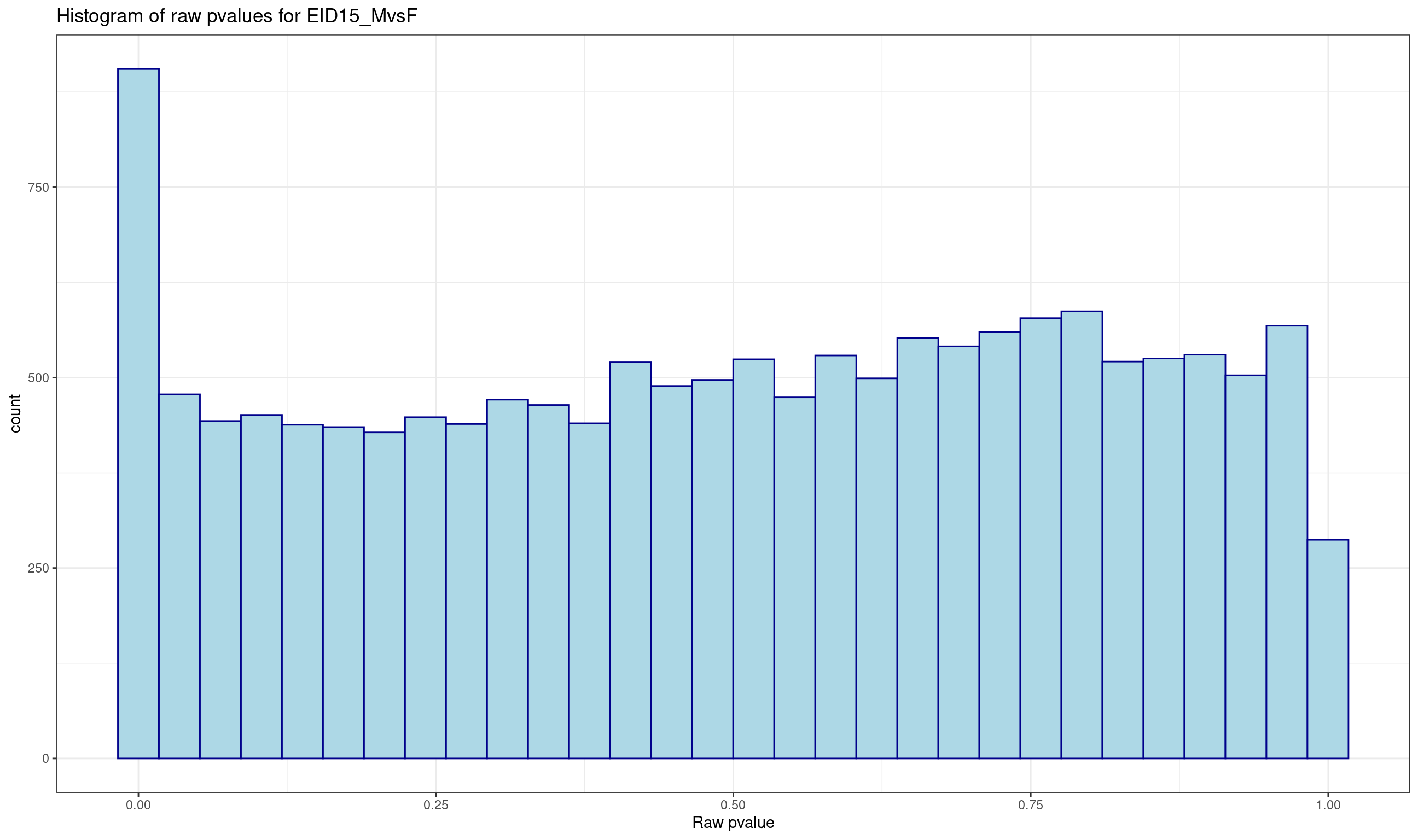
# MA plot
plotMD(lrt, main = "MA plot for EID15_MvsF")
Volcano plots represent a useful way to visualise the results of differential expression analyses. The x-axis is log2 fold change and represents biological significance. The y-axis is -Log10 Pvalue of the contrast tested and represents statistical significance. The highest genes on the graph are the most significantly differentially expressed (DE) genes.
See also the EnhancedVolcano
# volcano plot
p <- ggplot(data = res, aes(x = logFC, y = -log10(FDR)))
p <- p + geom_point(size=2, shape=19)
p <- p + ggtitle("Volcanoplot: Difference between males and females at EID15")
p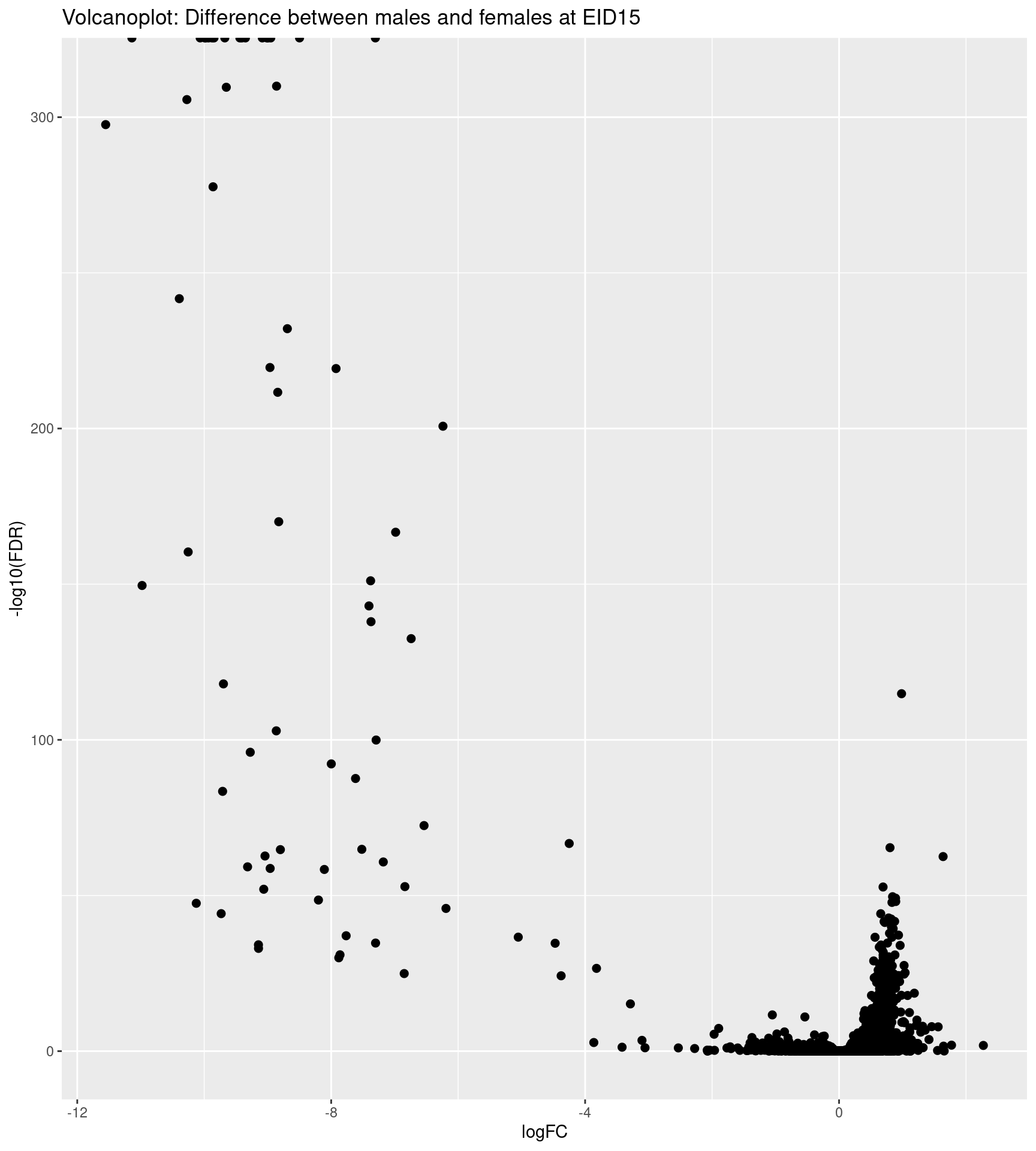
Save the results in separated files using a comma for the decimal point and a semicolon for the separator between columns.
# complete - all genes after filtering step
EID15_MvsF_bg <- res
write.csv2(EID15_MvsF_bg, file="output/complete_EID15_males_vs_females.csv")
# up regulated DE genes at FDR threshold 0.05
EID15_MvsF_up <- dplyr::filter(res, FDR <= 0.05 & logFC > 0)
write.csv2(EID15_MvsF_up, file="output/up_EID15_males_vs_females.csv")
# down regulated DE genes at FDR threshold 0.05
EID15_MvsF_down <- dplyr::filter(res, FDR <= 0.05 & logFC < 0)
write.csv2(EID15_MvsF_down, file="output/down_EID15_males_vs_females.csv")Functional enrichment test
The comparison of biological processes (BP) between the lists of DE genes were performed using ViSEAGO
Genes of interest
The results from differential analysis were used.
# Use a common background for EID11 et EID15 analyses
background <- unique(EID11_MvsF_bg$gene_id, EID15_MvsF_bg$gene_id)
DE_EID11_MvsF_up <- EID11_MvsF_up$gene_id[stringr::str_starts(EID11_MvsF_up$gene_id, "ENSGALG")]
DE_EID11_MvsF_down <- EID11_MvsF_down$gene_id[stringr::str_starts(EID11_MvsF_down$gene_id, "ENSGALG")]
DE_EID15_MvsF_up <- EID15_MvsF_up$gene_id[stringr::str_starts(EID15_MvsF_up$gene_id, "ENSGALG")]
DE_EID15_MvsF_down <- EID15_MvsF_down$gene_id[stringr::str_starts(EID15_MvsF_down$gene_id, "ENSGALG")]GO annotation of genes
The genes were previously annotated using biomaRt
# load GO annotations from Ensembl
load("./cam-data/CAM_myGENE2GO_Ensembl.RData")
myGENE2GO_Ensembl- object class: gene2GO
- database: Ensembl
- stamp/version: genes Ensembl Genes 106
- organism id: ggallus_gene_ensembl
GO annotations:
- Molecular Function (MF): 12702 annotated genes with 41502 terms (3406 unique terms)
- Biological Process (BP): 12653 annotated genes with 75167 terms (9994 unique terms)
- Cellular Component (CC): 12096 annotated genes with 43282 terms (1501 unique terms)Functionnal GO enrichment tests
At this step, we used myGENE2GO directly from Ensembl query. For each list of genes of interest, the GO enrichment tests were performed for BP category with Fisher’s exact test based on a common background of genes (all genes expressed in the study). The number minimal of genes is set to the value of nodeSize to consider valuable GO term.
# create topGOdata for BP
BP_EID11_MvsF_up <-ViSEAGO::create_topGOdata(
geneSel = DE_EID11_MvsF_up,
allGenes = background,
gene2GO = myGENE2GO_Ensembl,
ont = "BP",
nodeSize = 5
)
BP_EID11_MvsF_down <-ViSEAGO::create_topGOdata(
geneSel = DE_EID11_MvsF_down,
allGenes = background,
gene2GO = myGENE2GO_Ensembl,
ont = "BP",
nodeSize = 5
)
BP_EID15_MvsF_up <-ViSEAGO::create_topGOdata(
geneSel = DE_EID15_MvsF_up,
allGenes = background,
gene2GO = myGENE2GO_Ensembl,
ont = "BP",
nodeSize = 5
)
BP_EID15_MvsF_down <-ViSEAGO::create_topGOdata(
geneSel = DE_EID15_MvsF_down,
allGenes = background,
gene2GO = myGENE2GO_Ensembl,
ont = "BP",
nodeSize = 5
)# perform TopGO test using clasic algorithm
classic_EID11_MvsF_up <-topGO::runTest(
BP_EID11_MvsF_up,
algorithm ="classic",
statistic = "fisher"
)
classic_EID11_MvsF_down <-topGO::runTest(
BP_EID11_MvsF_down,
algorithm ="classic",
statistic = "fisher"
)
classic_EID15_MvsF_up <-topGO::runTest(
BP_EID15_MvsF_up,
algorithm ="classic",
statistic = "fisher"
)
classic_EID15_MvsF_down <-topGO::runTest(
BP_EID15_MvsF_down,
algorithm ="classic",
statistic = "fisher"
)
classic_EID11_MvsF_up
Description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
Ontology: BP
'classic' algorithm with the 'fisher' test
6416 GO terms scored: 17 terms with p < 0.01
Annotation data:
Annotated genes: 9851
Significant genes: 293
Min. no. of genes annotated to a GO: 5
Nontrivial nodes: 3115 classic_EID11_MvsF_down
Description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
Ontology: BP
'classic' algorithm with the 'fisher' test
6416 GO terms scored: 23 terms with p < 0.01
Annotation data:
Annotated genes: 9851
Significant genes: 84
Min. no. of genes annotated to a GO: 5
Nontrivial nodes: 1553 classic_EID15_MvsF_up
Description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
Ontology: BP
'classic' algorithm with the 'fisher' test
6416 GO terms scored: 27 terms with p < 0.01
Annotation data:
Annotated genes: 9851
Significant genes: 292
Min. no. of genes annotated to a GO: 5
Nontrivial nodes: 3205 classic_EID15_MvsF_down
Description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
Ontology: BP
'classic' algorithm with the 'fisher' test
6416 GO terms scored: 15 terms with p < 0.01
Annotation data:
Annotated genes: 9851
Significant genes: 65
Min. no. of genes annotated to a GO: 5
Nontrivial nodes: 1031 Combine enriched GO terms
The results of enrichment tests were combined in an interactive table. This table contains for each enriched GO terms, additional columns including the list of significant genes and frequency (ratio between the number of significant genes and number of background genes in a specific GO tag) evaluated by comparison.
BP_sResults<-ViSEAGO::merge_enrich_terms(
Input=base::list(EID11_MvsF_down=base::c("BP_EID11_MvsF_down","classic_EID11_MvsF_down"),
EID15_MvsF_down=base::c("BP_EID15_MvsF_down","classic_EID15_MvsF_down"),
EID11_MvsF_up=base::c("BP_EID11_MvsF_up","classic_EID11_MvsF_up"),
EID15_MvsF_up=base::c("BP_EID15_MvsF_up","classic_EID15_MvsF_up")
)
)# display the merged table
ViSEAGO::show_table(BP_sResults)# print the merged table in a file
ViSEAGO::show_table(BP_sResults,"./output/BP_sResults.xls")Graphs of GO enrichment tests
# count significant (or not) pvalues by condition
ViSEAGO::GOcount(BP_sResults)GO terms Semantic Similarity
The Semantic Similarity (SS) between enriched GO terms were computed using Wang’s method based on the topology of GO graph structure.
# initialyse
myGOs<-ViSEAGO::build_GO_SS(gene2GO=myGENE2GO_Ensembl,enrich_GO_terms=BP_sResults)
# compute all available Semantic Similarity (SS) measures
myGOs<-ViSEAGO::compute_SS_distances(myGOs,distance="Wang")Clustering heatmap of GO terms
A hierarchical clustering was performed using Wang SS distance between enriched GO terms and ward.D2 aggregation criteria. Enriched GO terms are ranked in a dendrogram and colored depending on their cluster assignation. A heatmap of -log10(pvalue) of enrichment test for each comparison and optionally the Information Content (IC) were displayed.
# GOterms heatmap with the default parameters
Wang_clusters_wardD2 <- ViSEAGO::GOterms_heatmap(
myGOs,
showIC=T,
GO.tree=base::list(
tree=base::list(
distance="Wang",
aggreg.method="ward.D2"
),
cut=base::list(
dynamic=base::list(
pamStage=T,
pamRespectsDendro=T,
deepSplit=2,
minClusterSize =4)
)
),
samples.tree=NULL
)# Display the clusters-heatmap
ViSEAGO::show_heatmap(Wang_clusters_wardD2, "GOterms", plotly_update = TRUE)# Display the clusters-heatmap table
ViSEAGO::show_table(Wang_clusters_wardD2)# Print the clusters-heatmap table
ViSEAGO::show_table(
Wang_clusters_wardD2,
"./output/cluster_heatmap_Wang_wardD2.xls"
)Multi Dimensional Scaling of GO terms
The MDS plot allows the user to visualise functional coherence as a whole and interactively explore functional subsets in greater detail.
# display colored MDSplot
ViSEAGO::MDSplot(Wang_clusters_wardD2)Results
This a summarise of the functional enrichment tests. Due to package updates since the publication, new enriched GO terms have appeared, resulting in slight differences in the heatmap and MDS plots.
print(BP_sResults)- object class: enrich_GO_terms
- ontology: BP
- method: topGO
- summary:
EID11_MvsF_down
BP_EID11_MvsF_down
description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
available_genes: 15124
available_genes_significant: 124
feasible_genes: 9851
feasible_genes_significant: 84
genes_nodeSize: 5
nodes_number: 6416
edges_number: 13970
classic_EID11_MvsF_down
description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
test_name: fisher
algorithm_name: classic
GO_scored: 6416
GO_significant: 23
feasible_genes: 9851
feasible_genes_significant: 84
genes_nodeSize: 5
Nontrivial_nodes: 1553
EID15_MvsF_down
BP_EID15_MvsF_down
description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
available_genes: 15124
available_genes_significant: 100
feasible_genes: 9851
feasible_genes_significant: 65
genes_nodeSize: 5
nodes_number: 6416
edges_number: 13970
classic_EID15_MvsF_down
description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
test_name: fisher
algorithm_name: classic
GO_scored: 6416
GO_significant: 15
feasible_genes: 9851
feasible_genes_significant: 65
genes_nodeSize: 5
Nontrivial_nodes: 1031
EID11_MvsF_up
BP_EID11_MvsF_up
description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
available_genes: 15124
available_genes_significant: 415
feasible_genes: 9851
feasible_genes_significant: 293
genes_nodeSize: 5
nodes_number: 6416
edges_number: 13970
classic_EID11_MvsF_up
description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
test_name: fisher
algorithm_name: classic
GO_scored: 6416
GO_significant: 17
feasible_genes: 9851
feasible_genes_significant: 293
genes_nodeSize: 5
Nontrivial_nodes: 3115
EID15_MvsF_up
BP_EID15_MvsF_up
description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
available_genes: 15124
available_genes_significant: 410
feasible_genes: 9851
feasible_genes_significant: 292
genes_nodeSize: 5
nodes_number: 6416
edges_number: 13970
classic_EID15_MvsF_up
description: Ensembl ggallus_gene_ensembl genes Ensembl Genes 106
test_name: fisher
algorithm_name: classic
GO_scored: 6416
GO_significant: 27
feasible_genes: 9851
feasible_genes_significant: 292
genes_nodeSize: 5
Nontrivial_nodes: 3205
- enrichment pvalue cutoff:
EID11_MvsF_down : 0.01
EID15_MvsF_down : 0.01
EID11_MvsF_up : 0.01
EID15_MvsF_up : 0.01
- enrich GOs (in at least one list): 51 GO terms of 4 conditions.
EID11_MvsF_down : 23 terms
EID15_MvsF_down : 15 terms
EID11_MvsF_up : 17 terms
EID15_MvsF_up : 27 termsReproducibility token
sessioninfo::session_info(pkgs = "attached")─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.4.1 (2024-06-14)
os Ubuntu 22.04.4 LTS
system x86_64, linux-gnu
ui X11
language (EN)
collate fr_FR.UTF-8
ctype fr_FR.UTF-8
tz Europe/Paris
date 2024-08-19
pandoc 3.1.11 @ /usr/lib/rstudio/resources/app/bin/quarto/bin/tools/x86_64/ (via rmarkdown)
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
AnnotationDbi * 1.66.0 2024-05-01 [1] Bioconductor 3.19 (R 4.4.0)
Biobase * 2.64.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.0)
BiocGenerics * 0.50.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.0)
dplyr * 1.1.4 2023-11-17 [1] CRAN (R 4.4.0)
edgeR * 4.2.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.0)
ggplot2 * 3.5.1 2024-04-23 [1] CRAN (R 4.4.0)
GO.db * 3.19.1 2024-05-21 [1] Bioconductor
graph * 1.82.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.0)
IRanges * 2.38.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.0)
kableExtra * 1.4.0 2024-01-24 [1] CRAN (R 4.4.0)
limma * 3.60.2 2024-05-19 [1] Bioconductor 3.19 (R 4.4.0)
magrittr * 2.0.3 2022-03-30 [1] CRAN (R 4.4.0)
S4Vectors * 0.42.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.0)
SparseM * 1.83 2024-05-30 [1] CRAN (R 4.4.0)
stringr * 1.5.1 2023-11-14 [1] CRAN (R 4.4.0)
topGO * 2.56.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.0)
ViSEAGO * 1.18.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.0)
[1] /home/chennequet/R/x86_64-pc-linux-gnu-library/4.4
[2] /usr/local/lib/R/site-library
[3] /usr/lib/R/site-library
[4] /usr/lib/R/library
──────────────────────────────────────────────────────────────────────────────References
1. Hennequet-Antier C, Halgrain M, Réhault-Godbert S. RNA-seq dataset of the chorioallantoic membrane of male and female chicken embryos, after 11 and 15 days of incubation. Data in Brief. 2024;110830. doi:https://doi.org/10.1016/j.dib.2024.110830.
2. Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology. 2010;11:R25. doi:10.1186/gb-2010-11-3-r25.
3. Dillies M-A, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, et al. A comprehensive evaluation of normalization methods for illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 2012;14:671–83.
4. Mc DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–97.
5. Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological). 1995;57:289–300. http://www.jstor.org/stable/2346101.
6. Brionne A, Juanchich A, Hennequet-Antier C. ViSEAGO: A bioconductor package for clustering biological functions using gene ontology and semantic similarity. BioData Mining. 2019;12:16. doi:10.1186/s13040-019-0204-1.
