
@M02944:130:000000000-ARV3J:1:1102:11673:1783 1:N:0:CACCGG
CTTGGTCATTTAGAG
+
***<<*AEF???***Analyser la qualité des données de séquençage Illumina
NGS
quality control
command line
L’objectif de ce tutoriel est d’analyser la qualité de données de séquençage Illumina avec seqkit
Introduction
Évaluer la qualité des données issues d’un séquençage haut-débit est la première étape à effectuer avant de se lancer dans des analyses bioinformatiques. Cela permet de déterminer si les données qui vous sont fournies sont conformes à ce que vous aviez demandé au prestataire de séquençage et si la qualité intrinsèque de ces données est suffisante. Ce tutoriel est spécifique aux données Illumina. Pour des données long-reads ou issus d’une autre technologie de séquençage, les outils peuvent être différents et les métriques interprétées différemment.
Note
Généralement, cette étape est effectuée par le prestataire de séquençage, mais mieux vaut vérifier par vous-même pour éviter les mauvaises surprises et pour commencer à vous familiariser avec vos données.
Les caractéristiques des données Illumina
Il est important de déterminer, entre autres :
- le nombre de reads pour chaque échantillon
- la qualité des reads
- leur longueur
- une éventuelle contamination
- la présence résiduelle d’adaptateurs de séquençage
Les données sont généralement fournies au format FASTQ. Si ce n’est pas le cas, il existe des outils de conversion.
Le format FASTQ
Le format FASTQ est le format de référence des reads bruts issus d’un séquençage Illumina. Il s’agit d’un fichier texte qui se compose d’informations relatives à chaque read. Ces informations sont regroupées sur 4 lignes.
Exemple d’une séquence de 15 nucléotides au format FASTQ :
Signification :
1. le nom de la lecture et des métadonnées propres au séquençage. Elle commence toujours par le caractère @
2. la séquence
3. un séparateur (caractère +) pouvant être suivi des mêmes informations que sur la ligne 1
4. les valeurs de qualité de chaque base de la lectureLes informations présentes sur la 1ère ligne sont indiquées dans la Table 1.
@<instrument>:<run number>:<flowcell ID>:<lane>:<tile>:<x-pos>:<y-pos>:<UMI> <read>:<is filtered>:<control number>:<index>| Element | Requirements | Description |
|---|---|---|
| @ | @ | Each sequence identifier line starts with @ |
| <instrument> | Characters allowed: a–z, A–Z, 0–9 and _ |
Instrument ID |
| <run number> | Numerical | Run number on instrument |
| <flowcell ID> | Characters allowed: a–z, A–Z, 0–9 | |
| <lane> | Numerical | Lane number |
| <tile> | Numerical | Tile number |
| <x_pos> | Numerical | X coordinate of cluster |
| <y_pos> | Numerical | Y coordinate of cluster |
| <UMI> | A/T/G/C/N | Optional, appears when UMI is specified in sample sheet. UMI sequences for Read 1 and Read 2, seperated by a plus [+] |
| <read> | Numerical | Read number. 1 can be single read or Read 2 of paired-end |
| <is filtered> | Y/N | Y if the read is filtered (did not pass), N otherwise |
| <control number> | Numerical | 0 when none of the control bits are on, otherwise it is an even number. On HiSeq X and NextSeq systems, control specification is not performed and this number is always 0. |
| <index> | A/T/G/C/N | Index of the read. |
Source : https://support.illumina.com/help/BaseSpace_OLH_009008/Content/Source/Informatics/BS/FileFormat_FASTQ-files_swBS.htm
Note
Pour des besoins spécifiques, vous pourriez avoir besoin d’utiliser ces informations (par exemple pour supprimer des reads selon leur position sur la flowcell en cas de problème technique).
La qualité des bases
La qualité de chaque base est encodée de façon à n’être représentée qu’avec un caractère, de façon à limiter la taille du fichier et faciliter sa compression. Cette ligne est difficilement interprétable pour un humain mais les logiciels sauront décoder l’information. Il existe différentes plages de qualité qui sont utilisées en fonction du type de séquençage effectué.
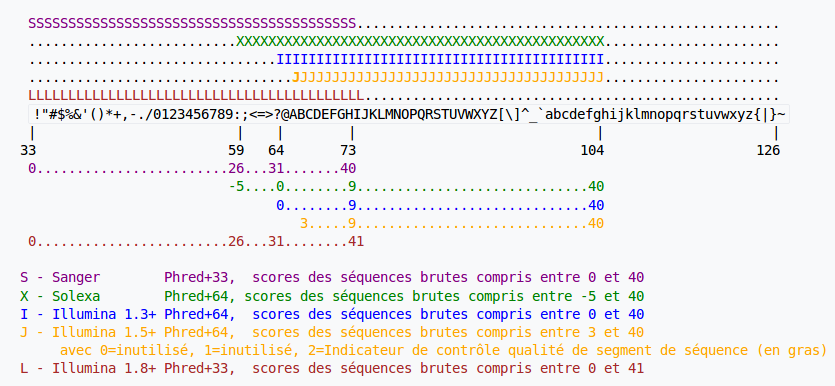
Note
Depuis 2011 et la sortie de Illumina 1.8, les plages de qualité sont uniformisées. Si vous avez des données produites avant 2011, il faudra peut être convertir la qualité pour quelle puisse être correctement interprétée par les outils.
Le score de qualité est lié de façon logarithmique à la probabilité d’erreur. Cette probabilité d’erreur est calculée lors de l’identification d’une base et correspond à la probabilité qu’il s’agisse d’une mauvaise identification.
Le taux d’erreur d’un séquençage Illumina est inférieur à 1% et dépend de la machine. Pour une revue très détaillée, vous pouvez consulter la publication de Stoler et Nekrutenko (2021) [4]: Sequencing error profiles of Illumina sequencing instruments
En fonction du type de données que vous allez traiter, il est possible que vous ayez envie/besoin de supprimer de vos reads des bases de mauvaise qualité (présentes souvent aux extrémités 3’ des reads). Cette étape s’appelle le trimming des reads et repose en partie sur l’information que vous aurez obtenue de l’analyse qualité.
Lire un fichier FASTQ
Un fichier FASTQ est très souvent compressé, ce qui permet de réduire énormément sa taille. En contrepartie, le contenu n’est plus lisible aussi facilement. Si vous avez besoin de lire un fichier FASTQ, pas pour tout regarder
Par exemple, pour afficher les 8 premières lignes d’un fichier FASTQ compressé :
zcat file.fastq.gz |head -n 8Pour rechercher le motif ATTCG dans le fichier :
zgrep ATTCG file.fastq.gz |headLes outils bioinformatiques
Il existe de nombreux outils bioinformatiques permettant d’analyser la qualité des données de séquençage. En fonction des technologies de séquençage utilisées pour générer les données, les informations importantes ne seront pas forcément identiques, et les outils non plus.
seqkit
seqkit
La sous-commande stat permet d’obtenir des informations générales sur le fichier analysé.
seqkit stat file.fastq.gzfile format type num_seqs sum_len min_len avg_len max_len
file1.fastq.gz FASTQ DNA 33,345 7,730,995 67 231.8 237Nous avons ici directement le nombre de reads, la longueur totale des reads, la taille minimale et maximale des reads ainsi que la longueur moyenne.
On peut bien sûr utiliser seqkit sur plusieurs fichiers :
seqkit stat *.fastq.gzfile format type num_seqs sum_len min_len avg_len max_len
file1.fastq.gz FASTQ DNA 33,345 7,730,995 67 231.8 237
file2.fastq.gz FASTQ DNA 33,345 7,800,533 68 233.9 238FastQC
FastQC
fastqc file.fastq.gzFastQC génère une archive file_fastqc.zip et un rapport HTML file_fastqc.html qu’il faut ouvrir avec un navigateur web :
firefox --no-remote file_fastqc.htmlVoici les informations que l’on peut obtenir en utilisant FastQC :
Per Base Sequence Quality
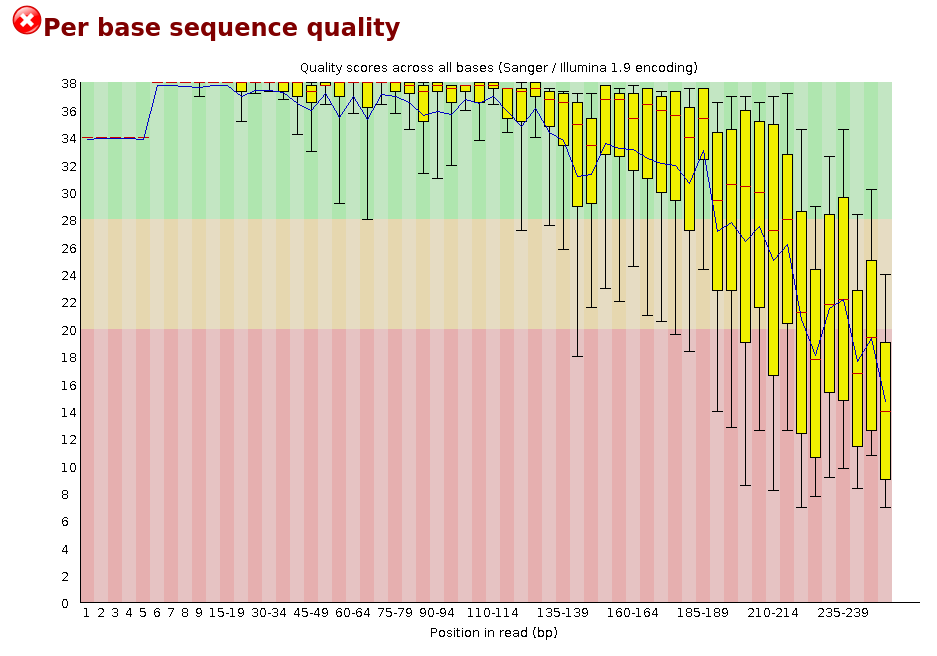
La Figure 2 représente la qualité (score Phred, en ordonnée) de chaque base (en abscisse) pour tous les reads de votre jeu de données. À chaque position du read, la qualité de tous les reads est représentée sous la forme d’un boxplot. La médiane est en rouge, la moyenne en bleu. Le code couleur vous indique les scores de très bonne qualité (en vert), bonne qualité (en orange) et mauvaise (en rouge). Généralement, la qualité baisse en fin de read. C’est ce graphique qui va vous indiquer par exemple si il faut trimmer vos reads, et à partir de quelle position le faire.
Per Sequence Quality Scores
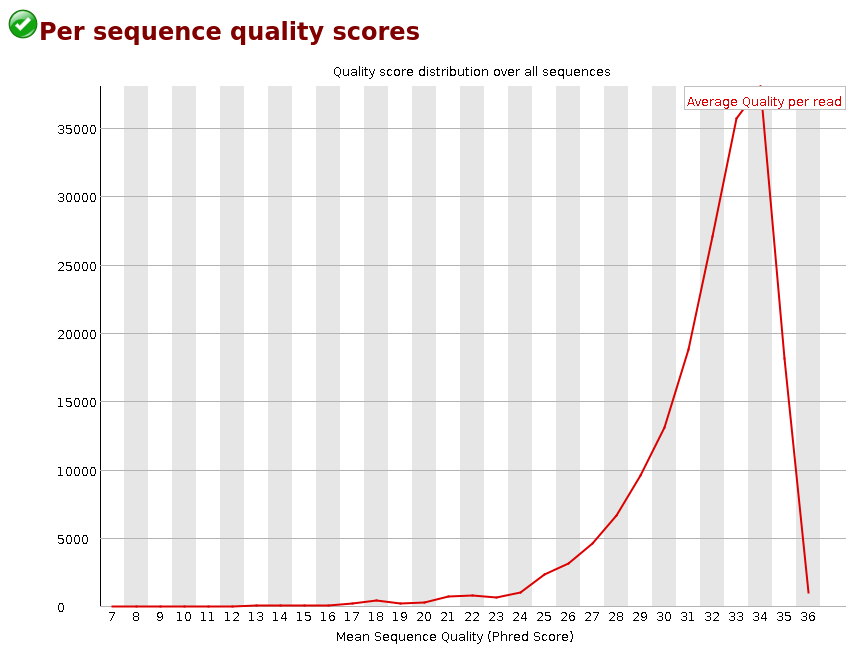
La Figure 3 représente la qualité moyenne par read. Cette qualité moyenne représente l’axe des abscisses et le nombre de reads ayant cette qualité est représenté sur l’axe des ordonnées. On s’attend à ce que la courbe soit haute pour les scores les plus élevés, comme c’est le cas ici. Ce graphique peut vous aider à trouver un seuil pour trimmer sur la qualité moyenne des reads, ce que certains outils de trimming proposent.
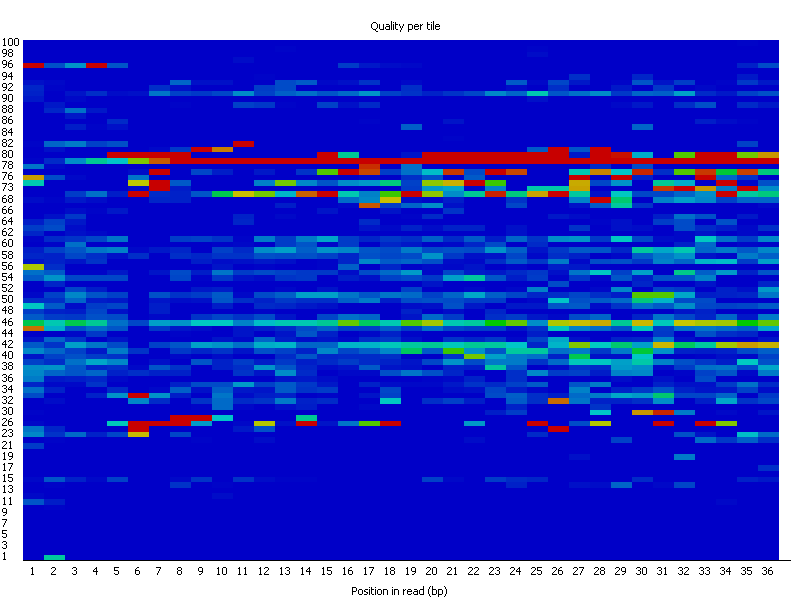
Per Tile Sequence Quality
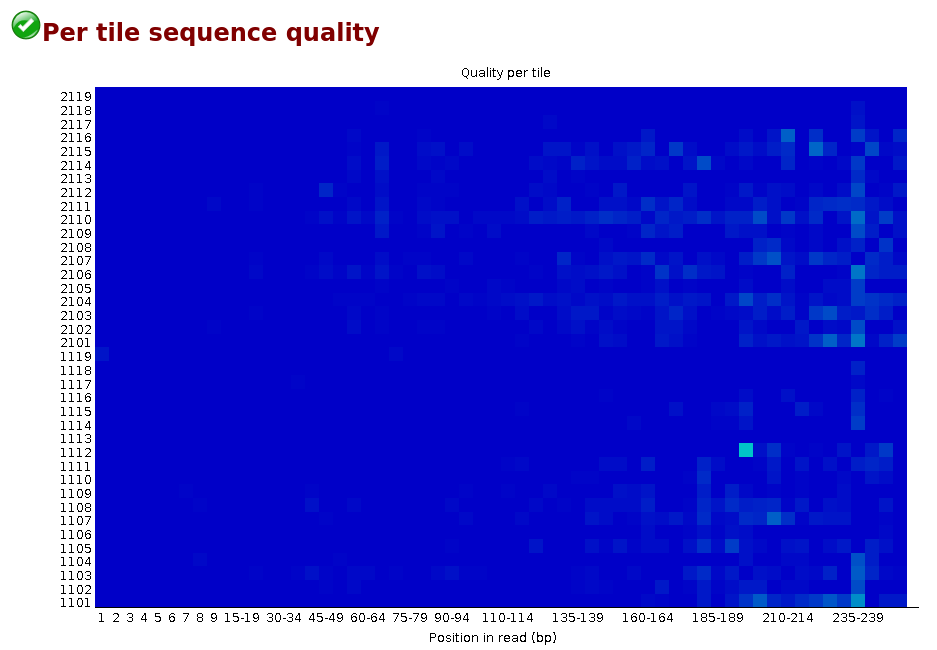
La Figure 4 est spécifique aux librairies Illumina. Chaque carré représente une partie de la flowcell. Cette information est présente dans l’identifiant de chaque read. Il permet de repérer si la qualité du séquençage est homogène sur la flowcell. Le graphique montre l’écart par rapport à la qualité moyenne pour chaque partie de la flowcell. Les couleurs sont sur une échelle de froid à chaud, les couleurs froides étant les positions où la qualité était égale ou supérieure à la moyenne pour cette base, et les couleurs plus chaudes indiquent une moins bonne qualité. Un graphique convenable devrait être bleu sur toute sa surface.
La Figure 5 un exemple de séquençage non homogène (donc problématique):
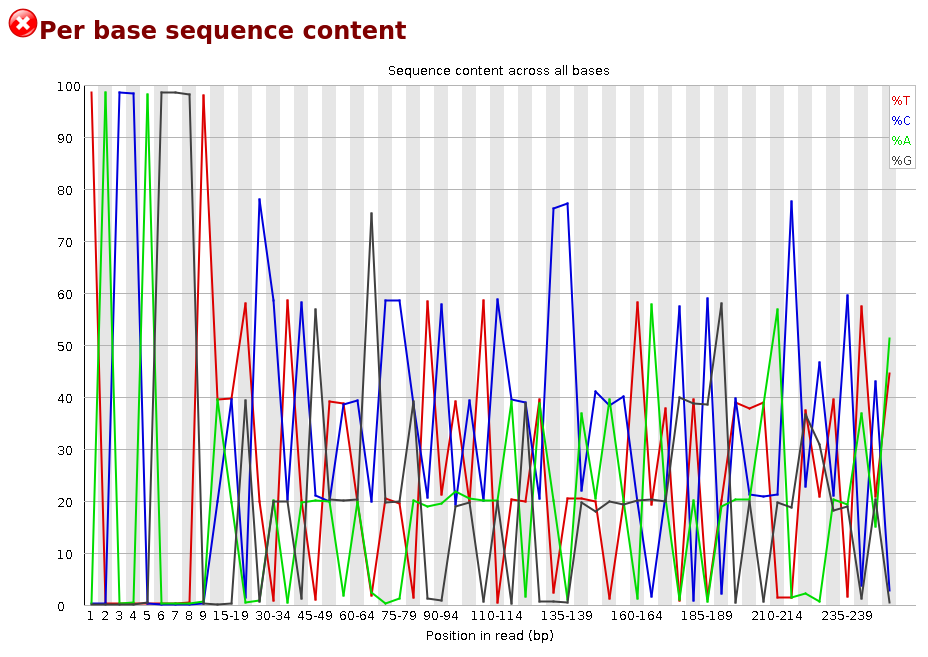
Per Base Sequence Content
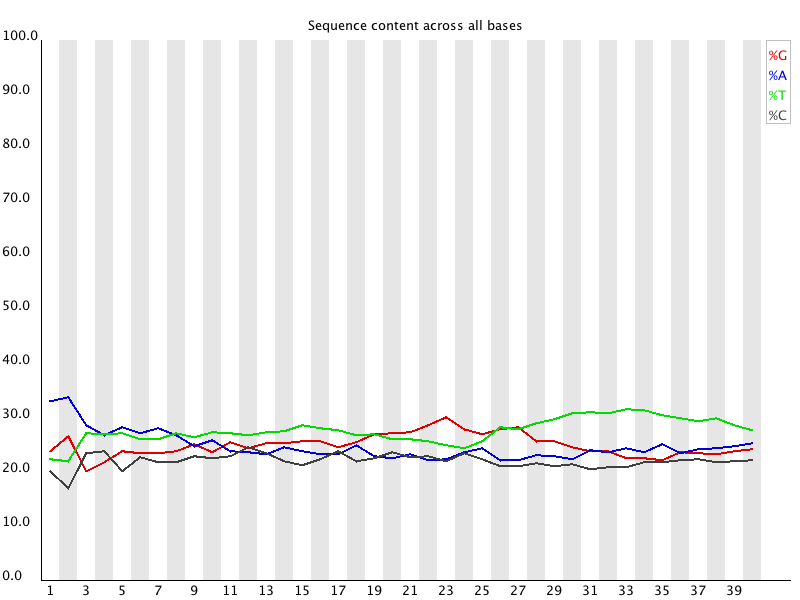
La Figure 6 représente le % de chaque base à chaque position du read. Dans une librairie aléatoire (random library), on s’attend à ce qu’il y ait peu ou pas de différence entre les différentes bases d’une séquence, de sorte que les lignes de ce graphique devraient être parallèles les unes aux autres. La quantité relative de chaque base devrait refléter la quantité globale de ces bases dans votre génome, mais dans tous les cas, elles ne devraient pas être très déséquilibrées les unes par rapport aux autres. Ce graphique n’est pas à regarder pour un séquençage d’amplicons !
La Figure 7 est caractéristique d’une librairie d’amplicons 16S.
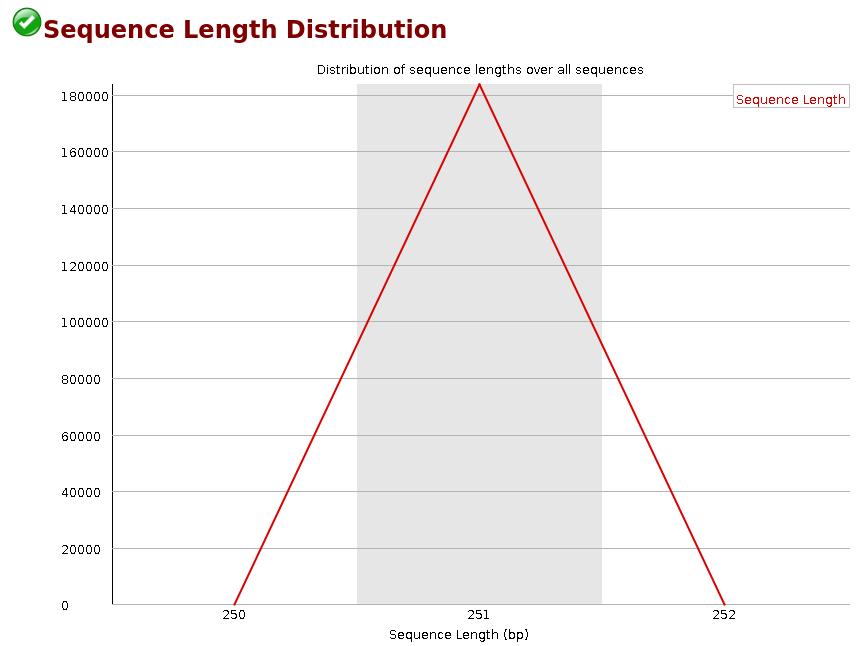
Sequence Length Distribution
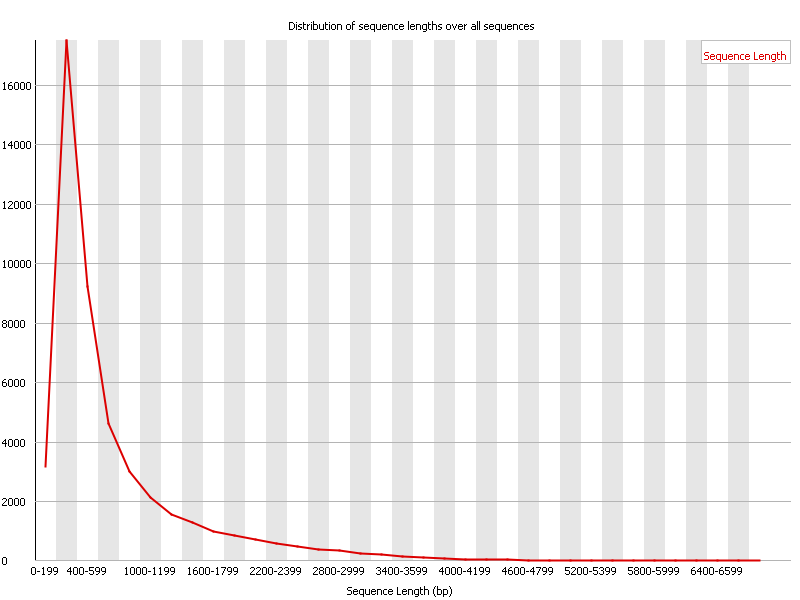
La Figure 8 représente la distribution des longueurs des reads. Il est inutile si vous vous attendez à avoir toutes les séquences de la même taille comme lors d’un séquençage Illumina. Il peut être très intéressant dans le cas d’une autre technologie (long-reads ou Ion Torrent) ou après un trimming
La Figure 9 est caractéristique d’une librairie d’amplicons 16S.
Per Sequence GC Content
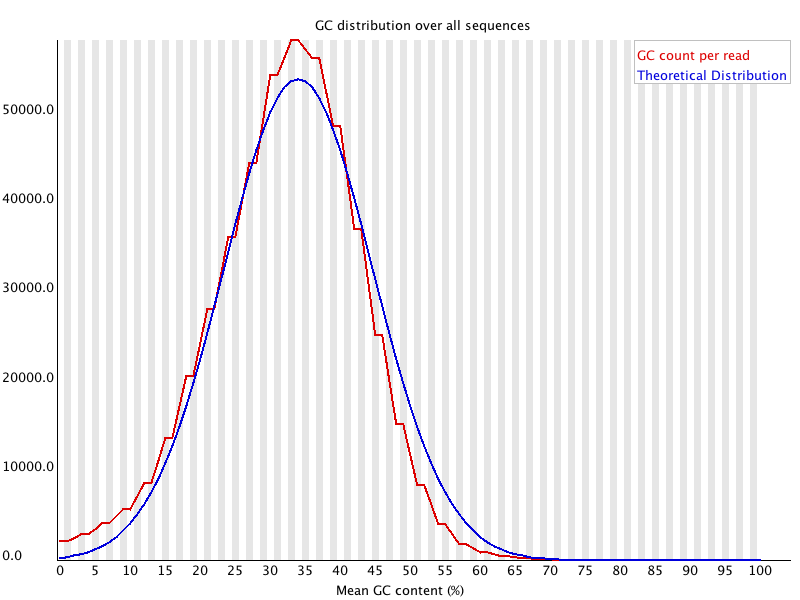
La Figure 10 représente le contenu en GC de chaque read et le compare à une distribution normale. Dans le cas d’un séquençage d’un seul organisme, ce graphique est très intéressant car il devrait refléter le contenu en GC attendu. Si ce n’est pas le cas, on peut suspecter une contamination ou un séquençage non aléatoire de la librairie. Dans d’autres cas de figure, ce graphique est inexploitable.
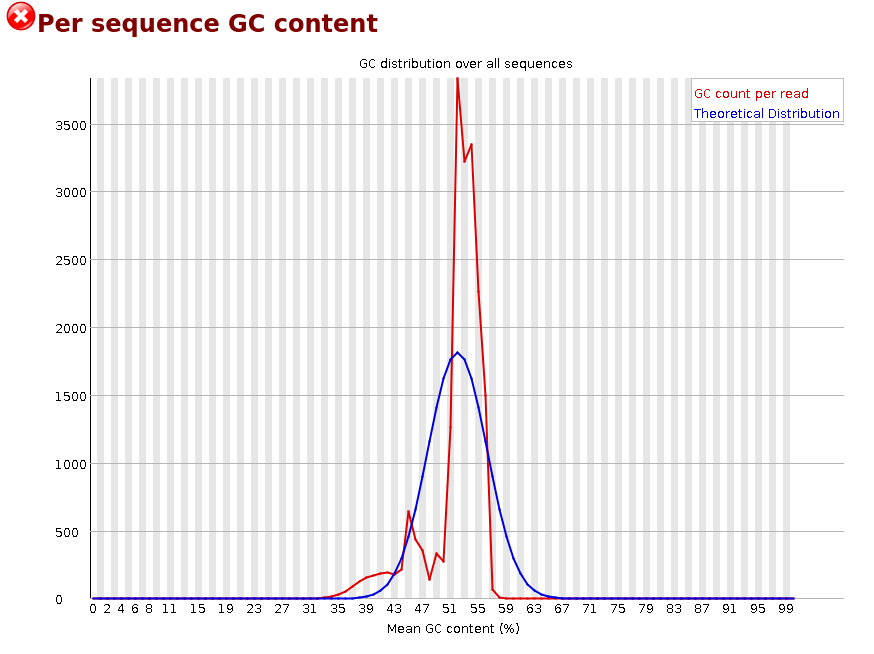
La Figure 11 est caractéristique d’une librairie d’amplicons 16S.
Per Base N Content
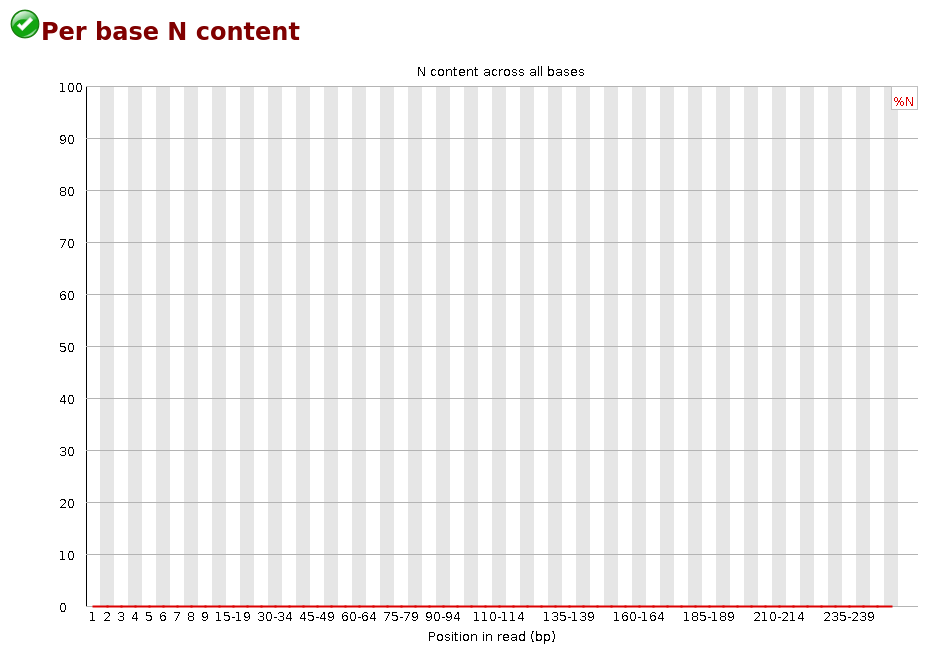
Si un séquenceur est incapable de déterminer la base (lors d’un cycle) avec suffisamment de confiance, il écrira un N plutôt qu’une base conventionnelle (A/C/G/T). La Figure 12 indique le pourcentage de N à chaque position. Généralement, très peu de N devraient être présents dans les reads si le séquençage s’est bien passé.
Adapter Content
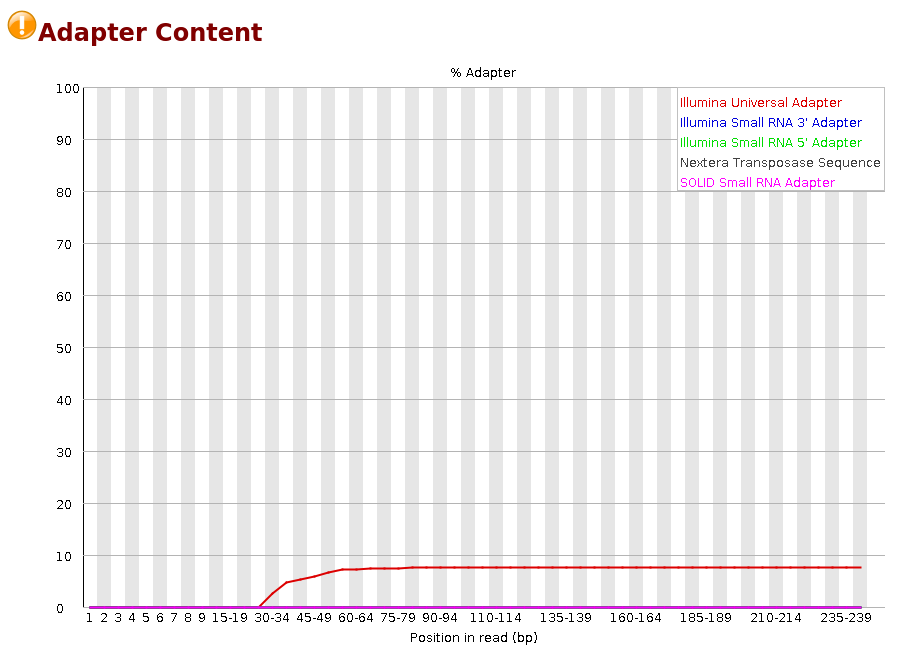
La Figure 13 permet de vérifier si les adaptateurs utilisés classiquement lors des séquençages sont présents dans les reads (par exemple les adaptateurs Illumina…). Si leur nombre est important, il faudra les retirer avec des outils dédiés (type sickle
MultiQC
MultiQC MultiQC, les bioinformaticiens regardaient (ou pas…) les rapports individuellement
Supposons que nous nous trouvions dans le répertoire contenant les rapports FastQC, il suffit de lancer MultiQC ainsi :
multiqc . -o MULTIQCUn rapport web est généré. Vous retrouverez une grande partie des graphiques présentés précédemment, mais pour tous les fichiers analysés ! La comparaison est alors très facile et déceler un fichier problématique est très rapide.
En voici un exemple sur un jeu de données amplicons 16S :
MultiQC va plus loin que résumer des sorties de FastQC. Il est capable de parser les logs générés par des centaines d’outils bioinformatiques.
Le site web de l’outil vous apportera toutes les informations utiles.
Mise en pratique sur l’infrastructure de Migale
Note
Tout ce qui suit peut être reproduit sur l’infrastructure migale. Les calculs sont bien entendus lancés sur les noeuds et non sur le frontal. Pour plus d’informations sur l’utilisation du cluster de calcul, voir le tutoriel dédié.
Le répertoire de travail sera ~/work/QC
ssh login@front.migale.inrae.fr # login
mkdir ~/work/QC
cd ~/work/QCRécupération de données
Nous allons récupérer trois jeux de données 16S déposés au NCBI et ayant pour identifiants SRR7127609, SRR7127610 et SRR7127614. Ce sont des données issues d’un séquençage Illumina Miseq, donc nous aurons deux fichiers par échantillon.
L’outil sra-tools
qsub -cwd -V -N download -b y "conda activate sra-tools-2.11.0 && fasterq-dump --split-files --progress --force --outdir . --threads 1 SRR7127609 SRR7127610 SRR7127614 && conda deactivate"Les données téléchargées n’étant pas compressées, nous allons nous empresser d’y remédier ! Vous verrez que la taille de chaque fichier sera divisée par 5 !
for i in *.fastq ; do gzip $i ; doneStatistiques avec seqkit
Nous allons maintenant lancer seqkit seqkit.tsv.
qsub -cwd -V -N seqkit -b y "conda activate seqkit-2.0.0 && seqkit stat *.fastq.gz > seqkit.tsv && conda deactivate"cat seqkit.tsv
file format type num_seqs sum_len min_len avg_len max_len
SRR7127609_1.fastq.gz FASTQ DNA 95,018 23,849,518 251 251 251
SRR7127609_2.fastq.gz FASTQ DNA 95,018 23,849,518 251 251 251
SRR7127610_1.fastq.gz FASTQ DNA 66,144 16,602,144 251 251 251
SRR7127610_2.fastq.gz FASTQ DNA 66,144 16,602,144 251 251 251
SRR7127614_1.fastq.gz FASTQ DNA 90,288 22,662,288 251 251 251
SRR7127614_2.fastq.gz FASTQ DNA 90,288 22,662,288 251 251 251Lancement de FastQC
Nous allons maintenant lancer FastQC FASTQC :
mkdir FASTQC
qsub -cwd -V -N fastqc -b y "conda activate fastqc-0.11.9 && fastqc *.fastq.gz -o FASTQC && conda deactivate"Vous pouvez ouvrir un rapport en utilisant firefox:
firefox --no-remote FASTQC/SRR7127609_1_fastqc.html &Lancement de MultiQC
Il vous reste à lancer MultiQC
qsub -cwd -V -N multiqc -b y "conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivate"Vous pouvez l’ouvrir avec firefox et l’analyser :
firefox --no-remote MULTIQC/multiqc_report.html &Conclusions
En savoir plus
Astuce
https://sequencing.qcfail.com/ recense les problèmes classiques liés au séquençage nouvelle génération.
Les références
1. Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PloS one. 2016;11:e0163962.
2. Andrews S. FastQC A Quality Control tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
3. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047‑8.
4. Stoler N, Nekrutenko A. Sequencing error profiles of Illumina sequencing instruments. NAR Genomics and Bioinformatics. 2021;3. doi:10.1093/nargab/lqab019.
5. Joshi N, Fass J. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files. 201apr. J.-C.
6. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10‑2. doi:10.14806/ej.17.1.200.
7. Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, et al. Database resources of the national center for biotechnology information. Nucleic acids research. 2006;35:D5‑12.
